Materials Genome through Data Mining
We are interested in accelerating the generation process of physical property databases by integrating a statistically-driven data mining protocol into a variety of modeling approaches including DFT-based electronic structure and molecular docking calculations.
Below are examples of our approaches:
Rapid acquisition on the mechanical properties of complex intermetallics
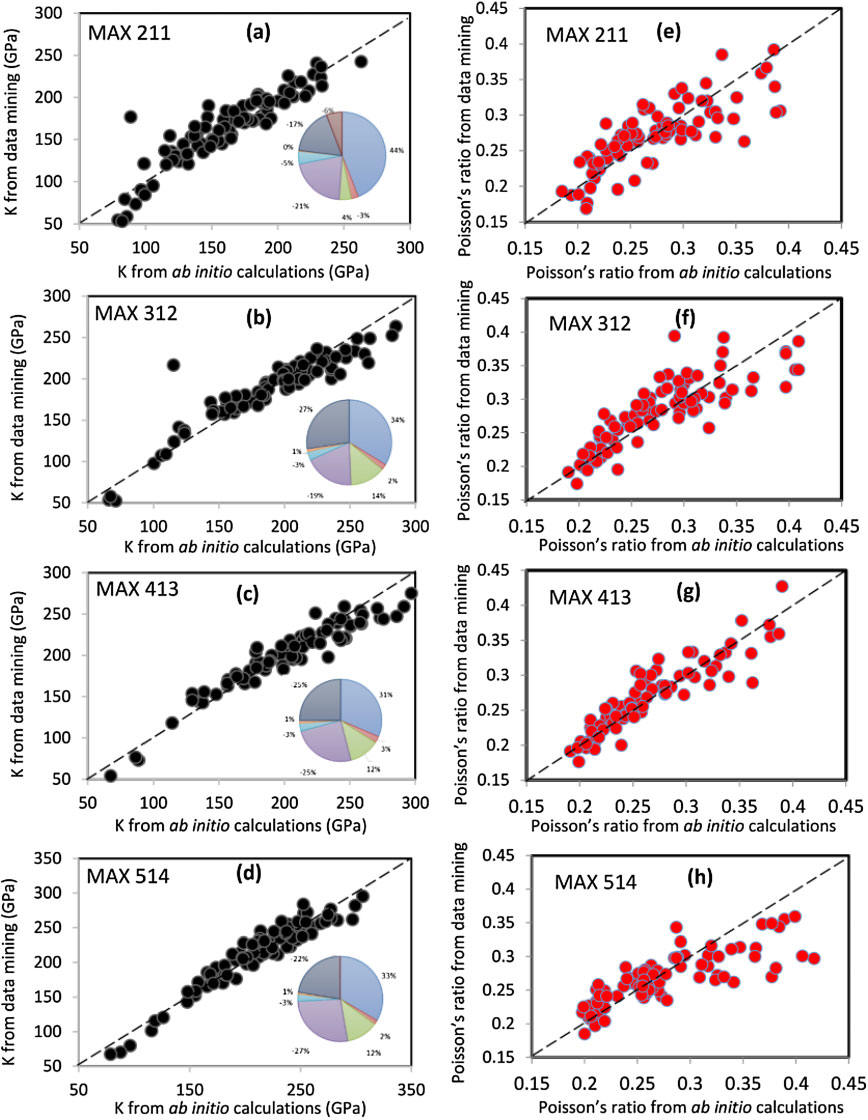
Here, we performed fundamental electronic structure calculations and we correlated them to more complex elastic properties. We employed the data mining tool as the means to bridge the results of the two types of calculations. We demonstrated the effectiveness of this approach to predict critical mechanical properties (bulk modulus and Poisson's ratio) of 655 ternary intermetallics called MAX phases (M= transition metals, A = Al/Ga/In/Tl/Si/Ge/Sn/Pb/P/As/S and X = C/N). The work was conducted as a part of collaborative work with Prof. W-Y Ching's research group at UMKC and Prof. Barsoum at Drexel University.
Rapid identification of critical rotatable bonds in molecular docking
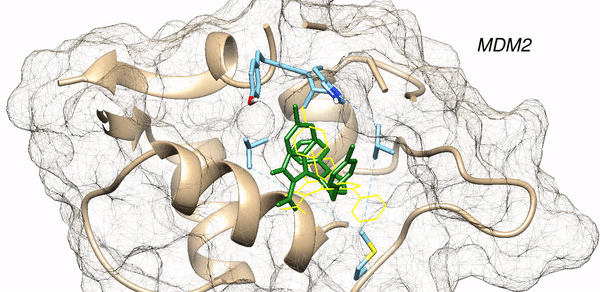
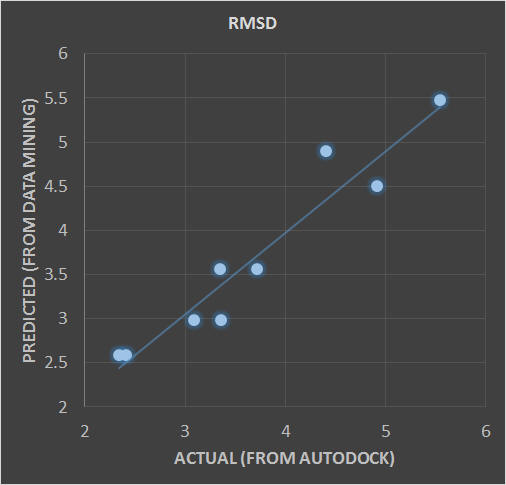
Given the large number of potential rotatable bond combinations between the ligand and protein, a ‘a priori’ knowledge of the optimal distribution of ligand/protein flexibility would be ideal considering testing all possible considerations would be impractical (in millions of possibilities). As a guide, we can employ a data mining approach where each of the potential bonds is then assigned as one of the parameters to be turned off or turned on (0 or 1). We can then seek the weight factor of each of these parameters by supposing the total cohesive energy as well as RMSD can be quantitatively linked to a linear regression as a superposition of all the parameters. Below is an example of such an application to correlate the total cohesive energy and RMSD obtained from the data mining approach with those obtained from more rigorous molecular docking methods using Autodock.
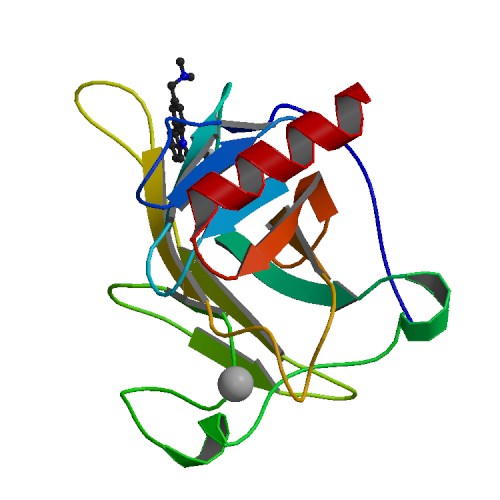
Docking results for structure 3LBK were used as a testing set for a linear regression analysis to determine the optimal flexibility distribution for the protein and ligand. Ideally, we want to isolate the critical rotatable bonds responsible for maximizing the magnitude of the total energy and/or minimizing the RMSD. In a way, it is a "genomic" approach, to ID the rotatable bonds. For 3LBK, overall we have identified P2, P4, P8 and P9 as the rotatable bonds that can be used to construct a number of simple linear models describing the values of RMSD and Total Energy (see the linear formulas below and the corresponding figure).
ENERGY = -1.4125 * P4 + 3.5027 * P8 - 1.8906 * P9 - 10.8333 (with protein only) CORR = 0.934
ENERGY = 2.1193 * L2 - 1.4588 * P2 + 3.8446 * P8 - 13.2877 (with protein & ligand) CORR= 0.953
RMSD = -1.3848 * P8 - 1.1181 * P9 + 3.7611 (with protein only) CORR = 0.777
RMSD = 1.7048 * L4 - 1.0319 * P8 + 1.6439 (with protein & ligand) CORR = 0.888
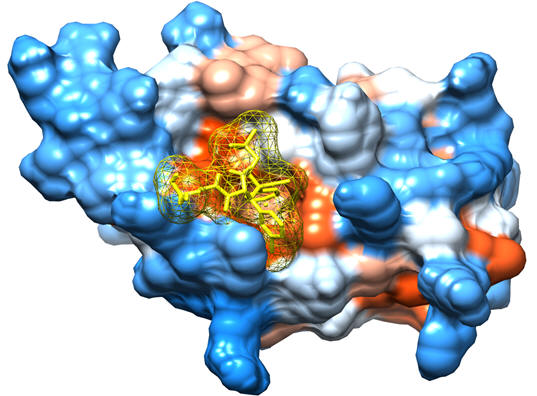
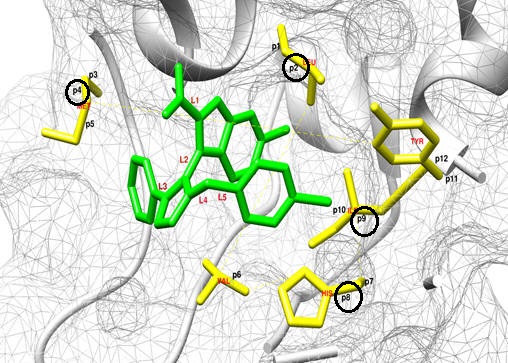
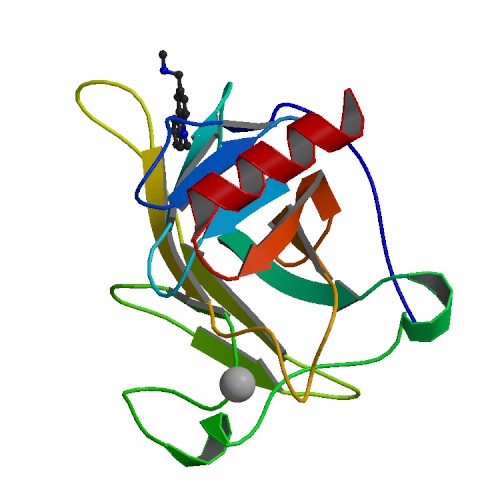
PDB Structure 3lbk with protein and ligand labels together with data mining analysis allows for a clear 3D assessment of the residues and rotatable bonds most critical for a successful docking.
More details can be found in our publication:
Anthony Ascone and Ridwan Sakidja, "MDM2 Case Study: Computational Protocol Utilizing Protein Flexibility Improves Ligand Binding Mode Predictions", International Journal of Computational Biology and Drug Design, Vol. 10 No. 3 pp. 217 - 224 (2017).
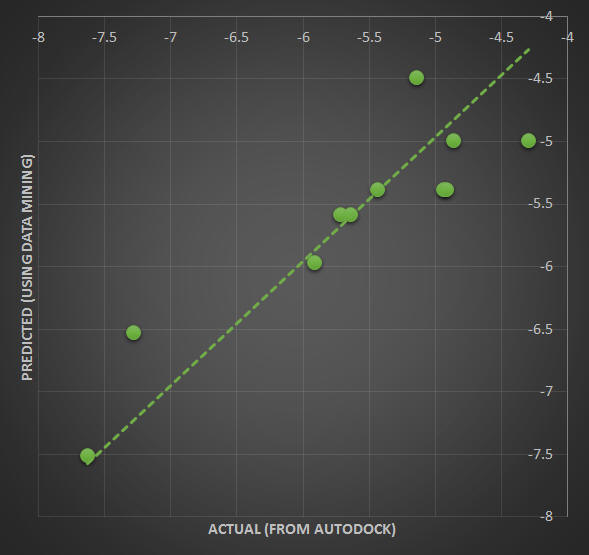
We have also applied a similar approach for 5G4O showing the interaction between the p53 cancer mutant Y220C in complex with a trifluorinated derivative of the small molecule stabilizer Phikan083. We identified an M5 pruned model tree with a correlation above 0.9 linking the total energy with the activation of specific rotatable bonds in the protein:
If P12 not active : LM#1 , if P12 active : LM#2
LM#1 : ENERGY = 0.9781 * P4 + 1.5416 * P9 - 7.5079
LM #2 : ENERGY = 1.0944 * P4 + 0.8892 * P9 + 0.524 * P12 - 6.9982
U
We identified a strong connection between the essential rotatable bonds and the RMSD:
RMSD = 0.5833 * L4 - 1.9117 * P8 + 0.98 * P9 + 3.9144
(CORRELATION = 0.96)