|
|
|
Computational methods have
advanced our understanding of bio-molecular binding processes and have become a
vital tool in structure-based drug design. Computational docking techniques in
particular, utilize free energy functions and algorithms, to rapidly produce a
quantitative ranking of likely binding sites of small molecules to larger target
molecules based on binding free energy. AutoDock is a widely used molecular
docking program that has been shown to be reasonably accurate predicting the
binding sites of ligands (small molecules) with 6 or fewer rotatable bonds.
However, accuracy decreases and becomes problematic after 10. We are interested
in finding ways to expedite this docking procedure. Our studies, which have
focused on protein flexibility, shows promise as the foundation for a new
screening protocol that would permit the accurate determination of binding sites
of larger ligands, while utilizing cost effective computational methods such as
the improved accuracy and speed of
Autodock Vina, a reliable new alternative to AutoDock.

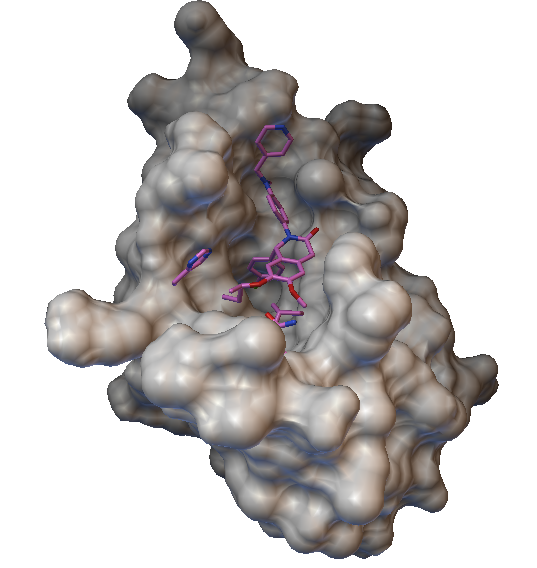
A snapshot from our research comparing the experimentally known structure (red) of
a cancer drug bound to the onco-protein MDM2
to AutoDock’s predicted binding site
In addition, we employ
accelerated Molecular dynamics simulation programs such as NAMD (Nanoscale Molecular
Dynamics) that use classical Newtonian physics to study the time dependant structure,
dynamics, and thermodynamics of biological molecules. The microscopic properties
of atomic positions and velocities can be translated into macroscopic quantities
such as temperature, pressure and volume using statistical mechanics. This
allows us to determine the flexible binding site residues of the target protein
most associated with these binding interactions. These side chain protein
residues can then be made flexible during docking runs in order to better
simulate changes in physical structure associated with ligand binding.

NVIDIA's GPU WORK STATION at MSU
Recenty, we have developed a simple approach to predict relevant rotatable bonds for a molecular docking. This approach allows us to quickly identify the pertinent residues from the protein that would greatly affect the effectiveness of the docking of screened drug candidates. See these examples in the Materials Genome section.