
![]()
2024
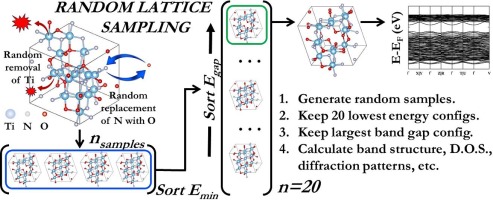
Abstract: The study presents the use of a novel on-lattice sampling approach to generate titanium oxynitride (TiNxOy) structures with potential applications in photovoltaic and water splitting. This approach presents a simple route to overcome challenges with structure-generating tools like Cluster Approach to Statistical Mechanics (CASM), and Ab initio Random Structure Search (AIRSS), CASM faces difficulty in generating ternary structures with large unit cells. With AIRSS, there is an increase in probability of sampling amorphous sample spaces with increased number of atoms in the unit cell. Here an on-lattice sampling approach was used to model the electronic structure of TiNxOy as a function of composition. We present results for Ti2N2O, Ti5N4O4 and Ti7N4O8, with 33 %, 50 % and 67 % N replaced by O via substitution relative to titanium nitride (TiN), respectively. Koopmans theorem was used correct the Kohn-Sham Density Functional Theory (KS-DFT) bandgaps with corresponding values of 2.68 eV, 3.03 eV, and 3.65 eV for 33, 50 and 67 % O doping respectively. The projected density of states (PDOS) plot for TiN shows that the Fermi level is dominated by the 3d atomic orbitals of Ti, confirming pure TiN’s metallicity. The valence bands of TiNxOy structures were dominated by 2p orbitals of O at lower energy levels, but they were dominated by 2p orbitals of N at energies close to the valence band maximum (VBM). The conduction bands were dominated by the 3d atomic orbitals of Ti, with the bandgap increasing with O composition leading to creation of shallow trap states near the VBM, which negatively impacts carrier mobility. In conclusion, the on-lattice sampling approach is an effective tool to generate highly crystalline structures of large unit cells, also keeping O substitution for N below 33 % as seen in Ti2N2O is crucial for avoiding shallow traps in TiNxOy structures.
2023

2023-3:
Andrew Ian Duff, Ridwan Sakidja, Helen C. Walker , Russell A. Ewings , David Voneshen, “Automated potential development workflow: application to BaZrO3", Computer Physics Communications, Volume 293, December 2023, 108896.

Abstract: We present a Python workflow, Automated Potential Development (APD), for automating the development of interatomic potentials, including calculation of density functional theory (DFT) fitting data, optimization of potentials, and potential-driven molecular dynamics (MD) simulations. The workflow currently supports CASTEP and VASP DFT codes and the MEAMfit potential optimization code for optimization of reference-free modified embedded atom method (RF-MEAM) potentials. The LAMMPS software is supported for calculating the relaxed geometry, elastic constants, phonon dispersion, thermal expansion and radial distribution functions using the optimized potentials. These same properties are also computed at the DFT level and APD automatically generates plots and tables of these data. Query-by-committee active learning is supported, using multiple fitted potentials to evaluate the energies of atomic configurations generated from LAMMPS MD runs. The workflow is demonstrated on BaZrO3, an oxide-based perovskite material, with RF-MEAM results found in good agreement with DFT.

Abstract: The electronic structure, interatomic bonding, and mechanical properties of two supercell models of Ni-based superalloys are calculated using ab initio density functional theory methods. The alloys, Haynes282 and Inconel740, are face-centered cubic lattices with 864 atoms and eleven elements. These multi-component alloys have very complex electronic structure, bonding and partial-charge distributions depending on the composition and strength of the local bonding environment. We employ the novel concept of total bond order density (TBOD) and its partial components (PBOD) to ascertain the internal cohesion that controls the intricate balance between the propensity of metallic bonding between Ni, Cr and Co, and the strong bonds with C and Al. We find Inconel740 has slightly stronger mechanical properties than Haynes282. Both Inconel740 and Haynes282 show ductile natures based on Poisson’s ratio. Poisson’s ratio shows marginal correlation with the TBOD. Comparison with more conventional high entropy alloys with equal components are discussed.
_____________________________________________________
2023-1: A. Tanji, R. Feng, Z. Lyu, R. Sakidja, Peter K. Liaw, H. Hermawan, "Passivity and corrosion resistance of Al20Cr5Fe50Mn20Ti5 and Al7Cr23.26Fe23.26Co23.26Ni23.26 high–entropy alloys in Hanks’ solution", Corrosion Science, Volume 210, Part 1, January 2023, 110828.

Abstract: The present work studies the passivation characteristic and corrosion resistance of Al20Cr5Fe50Mn20Ti5 and Al7Cr23.26Fe23.26Co23.26Ni23.26 high–entropy alloys in Hanks’ solution at 37 °C, in view of potential biomedical applications. The Al7Cr23.26Fe23.26Co23.26Ni23.26 possesses a higher corrosion resistance than Ti-6Al-4 V alloy, mainly due to the formation of a stable p–type passive film composed of Cr(OH)3, Cr2O3, CoO, Co3O4, and Ni(OH)2, leading to low vacancy concentration and limited ion conductivity. The Al20Cr5Fe50Mn20Ti5 exhibits an n–type passive film composed of Fe2O3, Fe(OH)3, TiO2, and MnO2 that promotes chloride ions adsorption and diffusion–controlled corrosion, while its microstructural precipitates allowed a selective dissolution.
_____________________________________________________
2022
2022-3: Ayoub Tanji, Rui Feng, Zongyang Lyu, Ridwan Sakidja, Peter K. Liaw, and Hendra Hermawan, "Niobium addition improves the corrosion resistance of TiHfZrNbx high-entropy alloys in Hanks’ solution", Electrochimica Acta, Vol. 424, pp. 140651(2022).
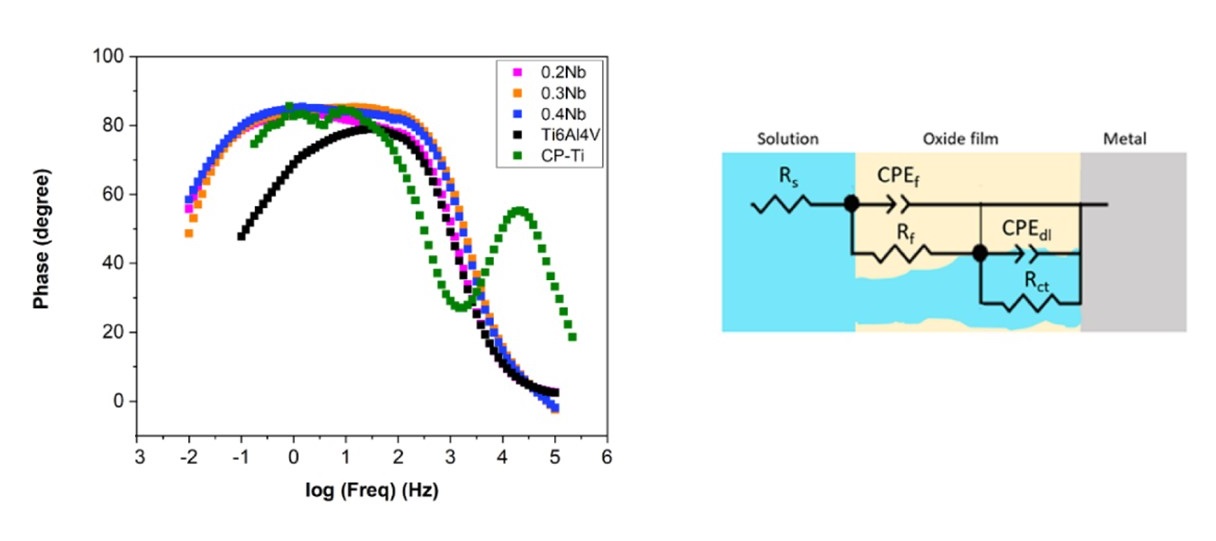
Abstract: High-strength TiHfZrNbx high-entropy alloys could become the ideal materials for small-diameter endovascular stents once their corrosion resistance in the physiological milieu is confirmed. This work aims at evaluating the corrosion resistance of a TiHfZrNbx high-entropy-alloys family in Hanks’ solution at 37 °C and revealing the influence of niobium on the mechanisms of dissolution and passivation. The alloys were subjected to a series of static and dynamic electrochemical tests and surface characterization, employing the static/dynamic/cyclic polarizations, impedance spectroscopy, XPS, AFM, and SEM. Results confirm a higher corrosion resistance of the alloys compared to that of CP-Ti and Ti6Al4V. The addition of niobium considerably improves the microstructural homogeneity that ensures a low dissolution rate and a greater resistance of the film due to the lower concentration of point defects. The passive film behaves as an n-type semiconductor and is composed of a mixture of TiO2, Nb2O5, ZrO2, and HfO2 oxides, with the presence of metallic hydroxides on the outermost layer. A detailed description of the niobium influence on the mechanisms of the dissolution and passivation is presented in this work.
_____________________________________________________
2022-2: Tyler McGilvry-James, Bikash Timalsina, Marium Mostafiz Mou, Ridwan Sakidja, "Deep potential development of transition-metal-rich carbides", MRS Advances, Vol. 7, pp. 468–473 (2022) , https://doi.org/10.1557/s43580-022-00289-0.
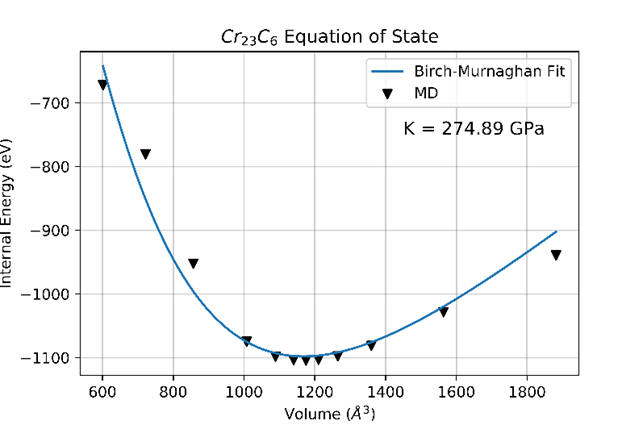
Abstract: In this study, we developed DeepPot-SE type potentials according to the High-Entropy Strategy for Cr23C6, Fe23C6, W23C6, and Mo23C6 systems. Predictive molecular dynamics are then conducted according to various elastic and mechanical properties. Training dataset accuracy is confrmed both numerically and visually. Elastic constants, Poisson Ratio, and bulk modulus are determined and compared to the literature with percent errors ranging between 0.45 and 13.33%. Ground-state lattice constants yield 0.1111.375% percent error. Linear lattice constant thermal expansion trends are found from 300 to 1500 K as expected. Melting behaviors with clear melting points are observed for each binary. The BirchMurnaghan Equation of State is suitably fit, providing additional verifcation of the bulk modulus calculations. Polycrystalline thermal stability is also verifed for each binary potential.
_____________________________________________________
2022-1: Devon Romine and Ridwan Sakidja, "Modeling atomic layer deposition of alumina using reactive force feld molecular dynamics", MRS Advances, Vol. 7, 185-189 (2022), https://10.1557/s43580-022-00289-0.
Abstract: In this study, we have utilized the reactive molecular dynamics (MD) simulations to model the Atomic Layer Deposition (ALD) process that forms an ultra-thin film of a tunnel barrier made of amorphous alumina. We chose the reactive MD approach over the ab-initio molecular dynamics simulation used in previous studies due to its lower computational cost, its ability to model over a relatively longer simulation period and its capability to assess atomistic-based dynamics for a larger substrate. We have reviewed the capabilities of ReaxFF to model stable precursors and reactions paths for two surface reactions of the ALD process utilizing LAMMPS and Amsterdam Modeling Suites software. A comparison of two force field potential models is also made in an effort to determine where deficiencies in the modeling capabilities lie.
_____________________________________________________
2021
2021-3: Maogang Gong, Bikash Timalsina, Ridwan Sakidja, Justin T. Douglas, Judy Z. Wu, "Ligands Anchoring Stabilizes Metal Halide Perovskite Nanocrystals", Advanced Optical Materials, Vol. 9, 2101012 (2021). https://doi.org/10.1002/adom.202101012
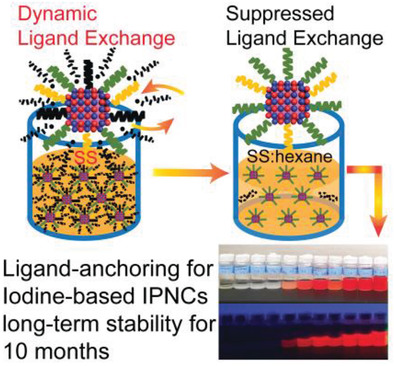
Abstract: Instability of colloidal iodine-based inorganic perovskite CsPbX3 (X = Cl, Br, I) nanocrystals (IPNCs) represents a major obstacle in lead-halide IPNC research and application. Herein, a ligand-anchoring process is reported that enables significantly improved colloidal stability of the iodine-based IPNCs for over 10 months in ambient. Apart from the previous efforts in searching for strong binding ligands to cap the IPNCs to incrementally reduce the exposure of the IPNC surface to the harsh colloidal environment, the ligand-anchoring method demonstrates that such an exposure can be reduced substantially by suppressing the dynamic ligand exchange around the colloidal IPNCs. In the IPNC synthesis solution with common oleic acid (OA) and oleylamine (OLA) ligands with relative weak binding to IPNCs, a systematic reduction of the ligand concentration using hexane by an order of magnitude has shown to be effective in achieving OA/OLA ligand-anchored iodine-based IPNCs with superior stability as confirmed in optical absorption, photoluminescence, 1H solution nuclear magnetic resonance spectroscopy, and photoresponse. This result has revealed that the intermittent exposure of the IPNC surface during the dynamic ligand exchange is a primary mechanism underlying the colloidal IPNC instability, which can be resolved in the ligand-anchoring process by suppressing such dynamic activities.
_____________________________________________________
2021-2: Khagendra Baral, Saro San, Ridwan Sakidja, Adrien Couet, Kumar Sridharan, and Wai-Yim Ching, "Temperature-Dependent Properties of Molten Li2BeF4 Salt Using Ab Initio Molecular Dynamics", ACS Omega, Vol. 6[30], 19822–19835, (2021).
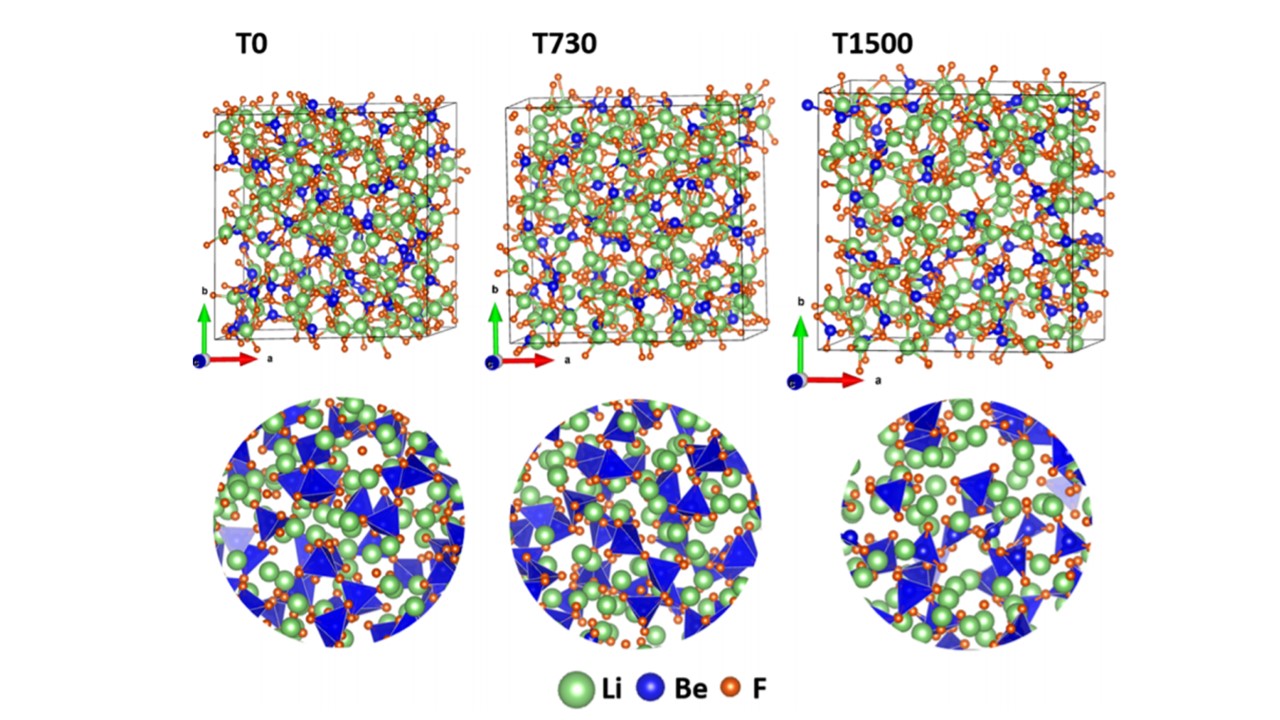
Abstract: Molten lithium tetrafluoroberyllate (Li2BeF4) salt, also known as FLiBe, with a 2:1 mixture of LiF and BeF2 is being proposed as a coolant and solvent in advanced nuclear reactor designs, such as the molten salt reactor or the fluoride salt cooled high-temperature reactor. We present the results on the structure and properties of FLiBe over a wide range of temperatures, 0–2000 K, from high-throughput ab initio molecular dynamics simulation using a supercell model of 504 atoms. The variations in the local structures of solid and liquid FLiBe with temperature are discussed in terms of a pair distribution function, coordination number, and bond angle distribution. The temperature-dependent electronic structure and optical and mechanical properties of FLiBe are calculated. The optical and mechanical property results are reported for the first time. The results above and below the melting temperature (~732 K) are compared with the experimental data and with data for crystalline FLiBe. The electronic structure and interatomic bonding results are discussed in correlation with the mechanical strength. A novel concept of total bond order density (TBOD), an important quantum mechanical parameter, is used to characterize the internal cohesion and strength in the simulated models. The results show a variation in the rate of change in properties in solid and liquid phases with anomalous behavior across the melting region. The observed trend is the decrease in mechanical strength, band gap, and TBOD in a nonlinear fashion as a function of temperature. The refractive index shows a surprising minimum at 850 K, among the tested temperatures, which lies above the melting point. These findings provide a new platform to understand the interplay between the temperature-dependent structures and properties of FLiBe salt.
_____________________________________________________
2021-1: W. C. Chang, S. Lee, C. -H, Chung, R. Sakidja and J. S. Park, "Ablation Stability of In Situ Al2O3 Layer in Aluminized AISI 4130 Steels under High-Temperature Plasma Flame Environments", Journal of Materials Engineering and Performance. https://doi.org/10.1007/s11665-021-05899-7.
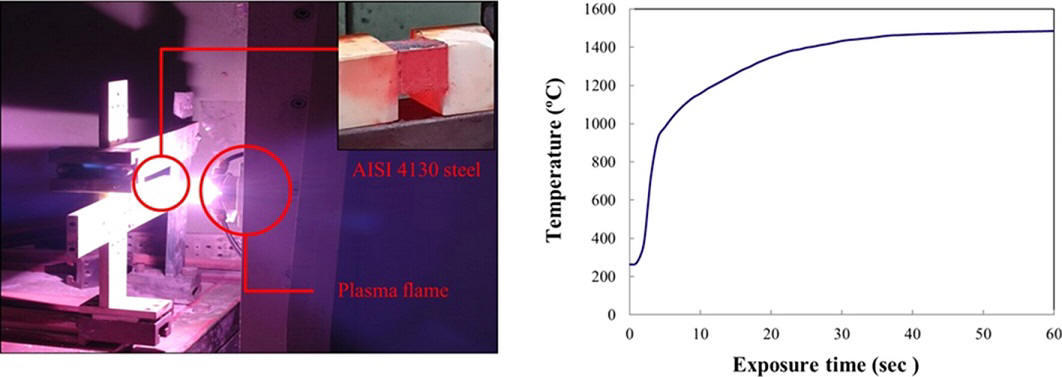
Abstract: The degradation and stability behavior of AISI 4130 steels have been examined under high-temperature dynamic plasma flame environments. A Fe2Al5 layer was formed as a result of aluminizing on the steel. An Al2O3 layer was produced on top of the Fe2Al5 layer, when the coated steel was oxidized at 500 °C (pre-oxidation treatment). The uncoated steels and the Fe2Al5 coated steels (i.e., the aluminized steels) showed large weight loss and complete changes in their original shapes due to their melting during the plasma flame tests at 1480 °C. However, the steels with the Al2O3 and Fe2Al5 (pre-oxidation treatment) were very stable without showing noticeable weight loss even after exposures to the plasma flame tests mainly due to the presence of the Al2O3 layer. SEM and XPS analyses were used to provide the presence of Fe2Al5 and Al2O3 phases on the surface. The effects of synthesized surface layers of the steels on high-temperature plasma flame exposure are discussed together with the surface morphology and microstructural observations.
_____________________________________________________
2020
2020-7: R. Goul, A. Marshall, R. Sakidja and J. Wu, "Investigation of in-vacuo atomic layer deposition of ultrathin MgAl2O4 using scanning tunneling spectroscopy", ACS Applied Electronic Materials, Vol. 2[10], 3121–3130 (2020).
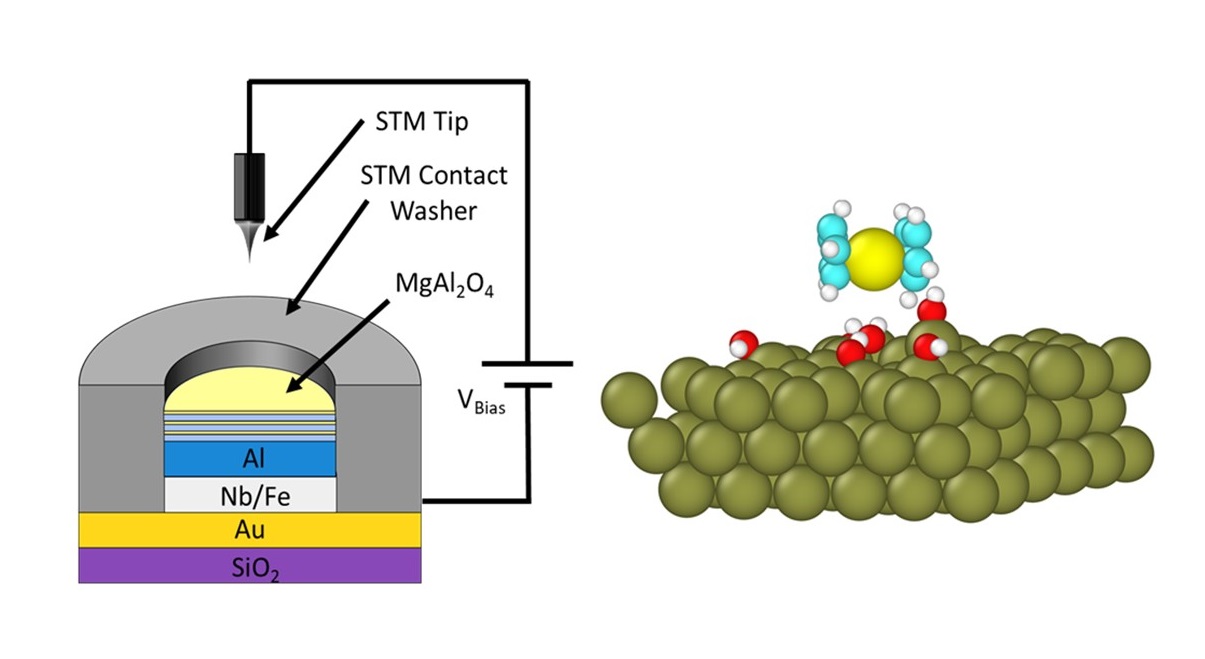
Abstract: Recently, disordered spinel MgAl2O4 as insulating tunnel barriers for perpendicular magnetic tunnel junctions has attracted interest due to their observed high tunneling magnetoresistance (TMR) and excellent voltage response. Motivated by this, we report the first success in the synthesis of ultrathin films (0.33–4.29 nm) of MgAl2O4 using in vacuo atomic layer deposition (ALD) on Fe and Al electrodes. The electronic properties of samples were evaluated using in situ scanning tunneling spectroscopy. Intriguingly, the sequence of the ALD Al2O3 and ALD MgO was found to dramatically impact the electronic structure of the ALD MgAl2O4, which may be attributed to the different initial adsorption mechanisms of ALD MgO and ALD Al2O3, as revealed in the molecular dynamics simulation. The optimum sequence for the first unit cell (or supercycle) of MgAl2O4 is two ALD Al2O3 cycles followed by one ALD MgO cycle. At three supercycles (0.99 nm), a much higher conduction band minimum (CBM) of 1.71 eV was observed, in contrast to 1.58 or 1.45 eV, which were observed when beginning the supercycles with 1 cycle of Al2O3 (0.11 nm) followed by 1 cycle of MgO (0.11 nm) or only 1 cycle of MgO, respectively. Decreasing the number of supercycles from 3 (~0.99 nm) to 1 supercycle (~0.33 nm) resulted in a monotonic decrease in CBM from 1.71 to 1.49 eV, showing some frustration of growth during earlier atomic layer deposition cycles. Additionally, growth on a Fe layer showed a moderate CBM of 1.25 eV. Nevertheless, the observed CBM in the ultrathin ALD MgAl2O4 greatly exceeds that of thermally oxidized AlOx barriers (~0.6 eV) and is similar to that of high-quality ALD-grown Al2O3 (~1.7 eV) and MgO grown with an Al2O3 seed layer (~1.50 eV) of comparable total thickness in the ultrathin range. The high CBM values are indicative of a low defect concentration in the ultrathin ALD MgAl2O4, which is supported by a high dielectric constant of 8.85 (comparable to that of the crystalline MgAl2O4 bulk) observed for a 4.3 nm thick ALD MgAl2O4 film capacitor.
_____________________________________________________
2020-6: M. Abbasi, S. Im, J. Johnson, G. Ortiz, M. Zhu, N. Oyler,M. Paquette, P. Rulis, R. Sakidja, J. Hwang, “Direct Determination of Medium Range Ordering in Amorphous Hydrogenated Boron Carbide for Low-k Dielectric Applications”, Microscopy and Microanalysis, Vol. 26 (Suppl 2), doi:10.1017/S143192762001394X (2020).
_____________________________________________________
2020-5: Wai-Yim Ching, Saro San, Jamieson Brechtl, Ridwan Sakidja, Miqin Zhang, and Peter Liaw, "Fundamental electronic structure and multiatomic bonding in thirteen biocompatible high-entropy alloys", npj Computational Materials, volume 6, Article number: 45 (2020).
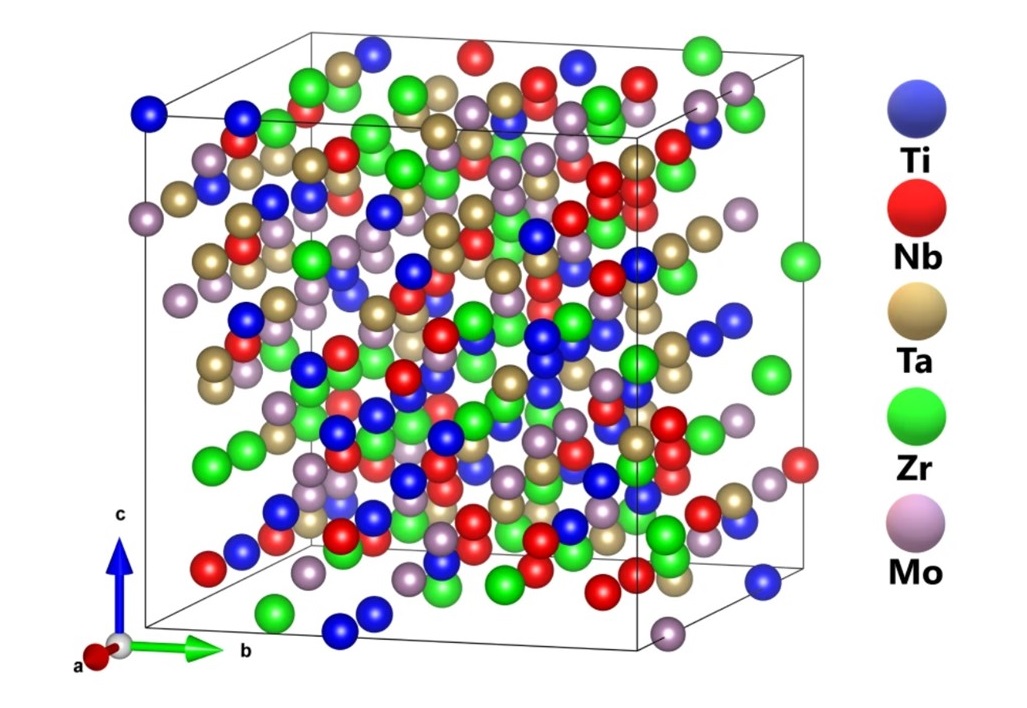
Abstract: High-entropy alloys (HEAs) have attracted great attention due to their many unique properties and potential applications. The nature of interatomic interactions in this unique class of complex multicomponent alloys is not fully developed or understood. We report a theoretical modeling technique to enable in-depth analysis of their electronic structures and interatomic bonding, and predict HEA properties based on the use of the quantum mechanical metrics, the total bond order density (TBOD) and the partial bond order density (PBOD). Application to 13 biocompatible multicomponent HEAs yields many new and insightful results, including the inadequacy of using the valence electron count, quantification of large lattice distortion, validation of mechanical properties with experiment data, modeling porosity to reduce Young’s modulus. This work outlines a road map for the rational design of HEAs for biomedical applications.
_____________________________________________________
2020-4: Nirmal Baishnab, Rajan Khadka, Michelle Paquette, Paul Rulis, Nathan Oyler, Jinwoo Hwang, Ridwan Sakidja, "Role of Generated Free Radicals in Synthesis of Amorphous Hydrogenated Boron Carbide from orthoCarborane using Argon Bombardment: A ReaxFF Molecular Dynamics Study", Materials Research Express, Vol. 6[12], 126461 (2020).

Abstract: In this study, we modeled and analyzed a critical aspect of the synthesis process for a-BxC:Hy i.e. the argon bombardment from the ortho-carborane precursor. Utilizing the reactive molecular dynamics simulations, we identified and evaluated the formation of free radicals as a result of ion bombardment. We found that increasing kinetic energy of ions releases an increasing amount of hydrogen. Thus, hydrogen content in the product can be tuned by varying the ion energy. Overall, our approach allows for a better understanding of the mechanism of Ar bombardment and the role of radical species toward the formation of ortho-carborane network.
_____________________________________________________
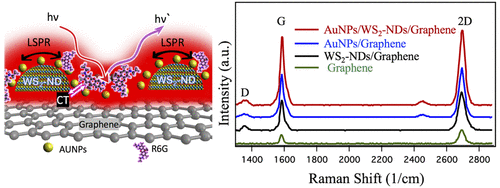
Abstract: This work explores superposition of the localized surface plasmonic resonance (LSPR) effect of Au nanoparticles (AuNPs) with that on transition metal dichalcogenide (TMD) WS2 nanodomes (WS2-NDs) enabled by enhanced dipole-dipole interaction at van der Waals (vdW) interfaces in AuNPs/WS2-NDs/graphene heterostructures for surface-enhanced Raman spectroscopy (SERS) with high-sensitivity, The confirmation of such a superposition is first demonstrated in the enhanced graphene Raman signatures, such as the G-peak intensity by approximately 7.8 fold on the AuNPs/WS2-NDs/graphene over that of reference graphene sample, in contrast to 4.0 and 5.3 folds respectively on AuNPs/graphene and on WS2-NDs/graphene. Furthermore, Raman spectra of probe molecules of fluorescent Rhodamine 6G (R6G) were hired to quantify the enhanced SERS on AuNPs/WS2-NDs/graphene SERS substrates. At the R6G concentration of 5x10-5 M, enhancement factors of ~ 2.0 and 2.4 based on the R6G 613 cm-1 peak intensity are detected on the AuNPs/WS2-NDs/graphene with respect to that on WS2-NDs/graphene and AuNPs/graphene, respectively. The benefit of the superposition of the LSPR effects from the WS2-NDs and AuNPs results in high SERS sensitivity up to 1×10-12 M on AuNPs/WS2-NDs/graphene, which is about an order of magnitude better than what’s on WS2-NDs/graphene, and several orders of magnitude better than that on the AuNPs/graphene and metal nanostructure/TMD (continuous layer) substrates. This result reveals the advantage of superposition of the LSPR effects from different nanostructures through design of vdW heterostructures. In addition, considering the AuNPs/WS2-NDs/graphene vdW heterostructures can be fabricated in the layer-by-layer growth developed in this work, the high-sensitivity SERS substrates are scalable and low cost for marketable devices in optoelectronics and biosensing.
_____________________________________________________
2020-2: Rajan Khadka, Nirmal Baishnab, George Opletal, Ridwan Sakidja "Study of amorphous Boron Carbide (a-BxC) materials using Molecular Dynamics (MD) and Hybrid Reverse Monte Carlo (HRMC)", J. Non-Crystalline Solids, Vol. 530, 119783 (2020).
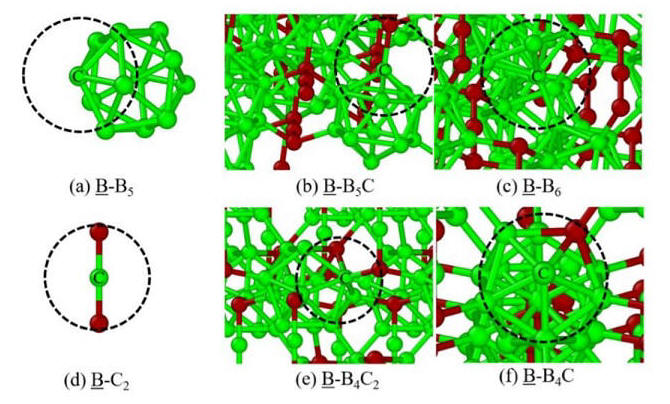
Abstract: We present a computational study of amorphous boron carbide (a-BxC) models using Molecular Dynamics (MD) studied with Stillinger-Weber (SW) and ReaxFF potential. The atomic structure factor (S(Q)), radial distribution function (RDF) and bond lengths comparison with other experimental and ab initio models show that a random arrangement of icosahedra (B12, B11C) interconnected by chains (CCC, CBC) are present in a-BxC. Subsequently, Hybrid Reverse Monte Carlo (HRMC) method is used to reconstruct a-BxC structures. The existing SW potential parameters of Boron are optimized for the α-rhombohedral (Icosahedral B12) boron structure using potential energy minimization and incorporated into HRMC. The a-BxC modeled from MD simulation is used as a sample for experimental input parameters like RDF, S(Q), coordination environments (CO), bond angle distribution (BAD) and bond length (BL) to guide initial configuration and simulation in HRMC. An accurate agreement of structural information between HRMC and MD generated models was found.
_____________________________________________________
2020-1: Tailei Qi, Youpin Gong, Alei Li, Xiaoming Ma, Peopei Wang, Rui Huang, Chang Liu, Ridwan Sakidja, Judy Z. Wu, Rui Chen, Liyuan Zhang, "Interlayer Transition in a vdW Heterostructure toward Ultrahigh Detectivity Shortwave Infrared Photodetectors", Advanced Functional Materials, 1905687, (2020).
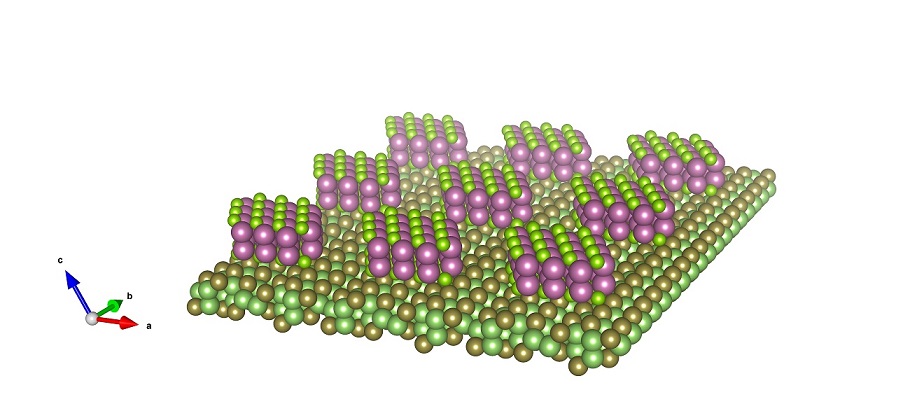
Abstract: Van der Waals (vdW) heterostructures of 2D atomically thin layered materials (2DLMs) provide a unique platform for constructing optoelectronic devices by staking 2D atomic sheets with unprecedented functionality and performance. A particular advantage of these vdW heterostructures is the energy band engineering of 2DLMs to achieve interlayer excitons through type-II band alignment, enabling spectral range exceeding the cutoff wavelengths of the individual atomic sheets in the 2DLM. Herein, the high performance of GaTe/InSe vdW heterostructures device is reported. Unexpectedly, this GaTe/InSe vdWs p–n junction exhibits extraordinary detectivity in a new shortwave infrared (SWIR) spectrum, which is forbidden by the respective bandgap limits for the constituent GaTe (bandgap of ≈1.70 eV in both the bulk and monolayer) and InSe (bandgap of ≈1.20–1.80 eV depending on thickness reduction from bulk to monolayer). Specifically, the uncooled SWIR detectivity is up to ≈1014 Jones at 1064 nm and ≈1012 Jones at 1550 nm, respectively. This result indicates that the 2DLM vdW heterostructures with type-II band alignment produce an interlayer exciton transition, and this advantage can offer a viable strategy for devising high-performance optoelectronics in SWIR or even longer wavelengths beyond the individual limitations of the bandgaps and heteroepitaxy of the constituent atomic layers.
2019
2019-8: Puja Adhikari, Saro San, Caizhi Zhou, Ridwan Sakidja, Wai-Yim Ching, "Electronic structure and mechanical properties of crystalline precipitate phases M23C6 (M = Cr, W, Mo, Fe) in Ni-based superalloys", Materials Research Express, Vol 6 [11], 116323, (2019).

Abstract: The presence of precipitate phases M23C6 (M = Cr, Fe, Mo, or W) plays a significant role in the mechanical strength of Ni-based superalloys. The crystal structure of these complex carbides consists of 116 atoms in the cubic cell of an fcc lattice. Here we report the investigation of the electronic structure, interatomic bonding and mechanical properties for four binary phases of M23C6, and M23N6, and six ternary phases of (M1, M2)23C6. Based on the results of our comprehensive calculations and the evaluation of a quantum mechanical metric called the total bond order density (TBOD), we conclude that Fe tends to make the intermetallic structure less stable whereas W makes the crystal more compressible. We trace the root of these observations to the size of the metal ions and the complex interatomic bonding between C atoms and the metal d electrons. In addition, we correlate the TBOD with calculated mechanical properties leading to confirmation that Cr23C6 is the most stable precipitate phase with superior overall physical properties. On the other hand, replacing C with N in these crystals does not result in discernable differences in their properties. Our detailed investigation sheds much light on the use of carbides precipitates as a stabilization strategy for Ni-based superalloys.
_____________________________________________________
2019-7: Jagaran Acharya, Ryan Goul, Devon Romine, Ridwan Sakidja, Judy Z. Wu, "Effect of Al2O3 Seed-Layer on the Dielectric and Electrical Properties of Ultrathin MgO Films Fabricated using In Situ Atomic Layer Deposition", ACS Applied Materials & Interfaces, v. 11 [33], pp. 30368-30375, (2019)
.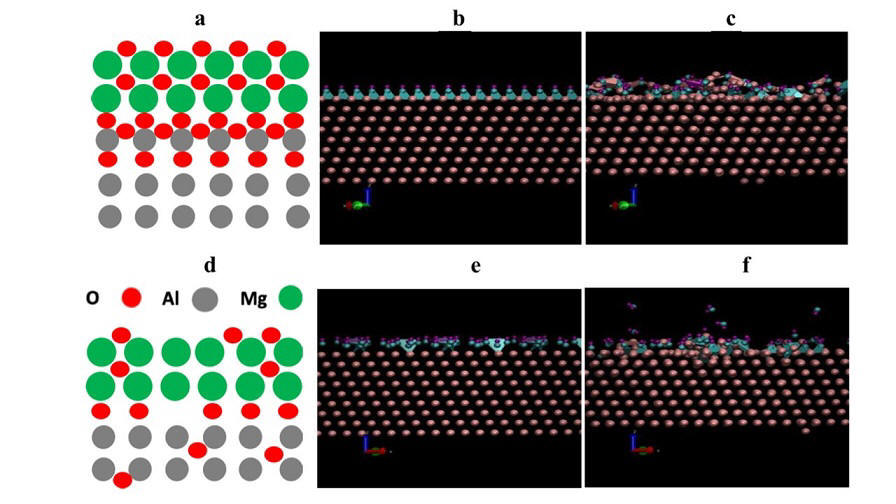
Abstract: Metal/insulator/metal (M/I/M) trilayers of Al/MgO/Al with ultrathin MgO in the thickness range of 2.20-4.40 nm were fabricated using in vacuo sputtering and atomic layer deposition (ALD). In order to achieve high-quality metal/insulator (M/I) interface and hence high-quality dielectric ALD-MgO films, a 5 cycles (~0.55 nm) thick ALD-Al2O3 seed layer (SL) was employed to demonstrate the dielectric constant (εr) ~8.82-9.38 in 3.30-4.95 nm thick ALD-MgO/SL films, which is close to that of single-crystal MgO εr ~9.80. In contrast, a low εr of 3.55-4.66 for the ALD-MgO films of similar thickness without a SL was observed. The effective oxide thickness (EOT) ~1.40 nm has therefore been achieved in the ultrathin ALD-MgO films, which are comparable to the EOTs of high-K dielectrics such as HfO2. In addition, the leakage current through the M/I/M structure is reduced by more than one order of magnitude with the implementation of the SL. The high leakage current in the samples without a SL can be attributed to the non-uniform nucleation of the ALD-MgO on the Al surface with a significant portion of the Al surface remaining conductive as confirmed using in vacuo scanning tunneling spectroscopy (STS). With the SL, the STS study has confirmed a tunnel barrier height of 1.50 eV on 0.55 nm MgO with 0.55 nm Al2O3 SL with almost 100% coverage. In addition, molecular dynamics simulations point out the importance of deposition of ultrathin SL that has a significant effect on the initial nucleation of Mg precursor. This result not only illustrates the critical importance of controlling M/I interface to obtain high quality dielectric properties of ultrathin ALD films but also provides an approach to engineering incompatible M/I interfaces using a SL for high quality dielectric required for applications in M/I/M tunnel junctions and complementary metal oxide semiconductor.
_____________________________________________________
2019-6: Tianju Chen, Ridwan Sakidja, Wai-Yim Ching, Caizhi Zhou, "Crystal Plasticity Modeling of Void Growth on Grain Boundaries in Ni-Based Superalloys", JOM, Volume 71, Issue 11, pp 3859–3868, (2019).
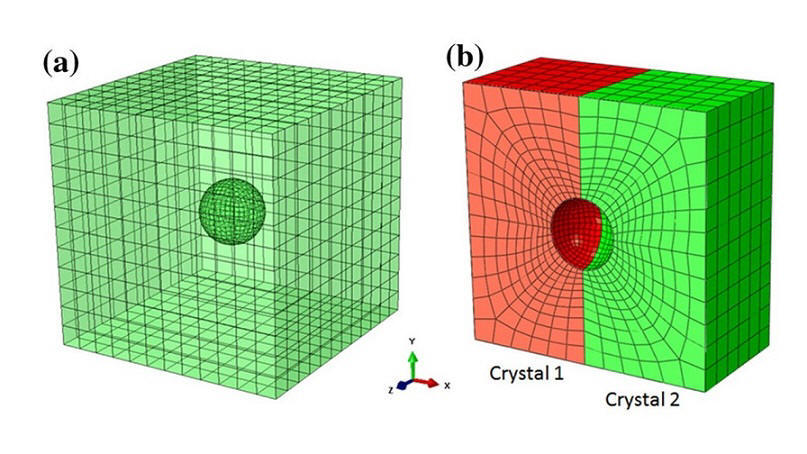
Abstract: Atomic interactions pertaining to carbon-nanocomposites can be elusive and hard to comprehend, and as such great benefit can be gained through a visualization of these interactions within a virtual or augmented reality setting. In this paper, we present a framework that can be used for Material Science research and education incorporating topics related to the dynamics in nanomaterials. We developed the proof of concept implementation of this framework for Virtual Reality (VR) and Augmented Reality (AR) settings using the Unity game engine. Throughout this paper, we discuss our framework as well as the related user experiences and performance measurements we gathered when using our framework with the Google Daydream, HTC Vive, and Microsoft HoloLens in introducing scientists to the use of AR and VR as a tool for nanocomposite and molecular dynamics research.
_____________________________________________________
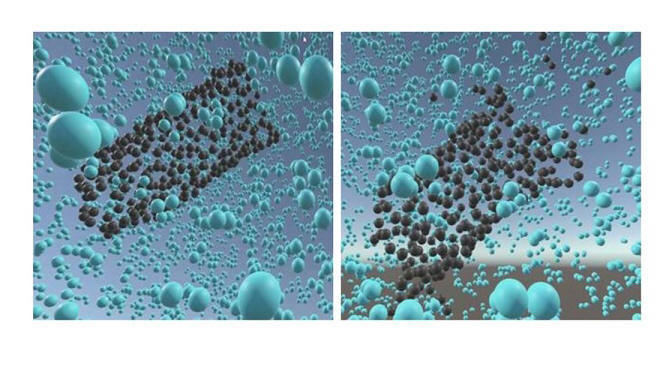
Abstract: Atomic interactions pertaining to carbon-nanocomposites can be elusive and hard to comprehend, and as such great benefit can be gained through a visualization of these interactions within a virtual or augmented reality setting. In this paper, we present a framework that can be used for Material Science research and education incorporating topics related to the dynamics in nanomaterials. We developed the proof of concept implementation of this framework for Virtual Reality (VR) and Augmented Reality (AR) settings using the Unity game engine. Throughout this paper, we discuss our framework as well as the related user experiences and performance measurements we gathered when using our framework with the Google Daydream, HTC Vive, and Microsoft HoloLens in introducing scientists to the use of AR and VR as a tool for nanocomposite and molecular dynamics research.
_____________________________________________________
2019-4: Mohammed Alamri, Ridwan Sakidja, Ryan Goul, Samar Ghopry, and Judy Z. Wu,"Plasmonic Au Nanoparticles on 2D MoS2/graphene van der Waals Heterostructures for High-Sensitivity Surface Enhanced Raman Spectroscopy", ACS Applied Nano Materials, 2 (3), 1412-1420, (2019)
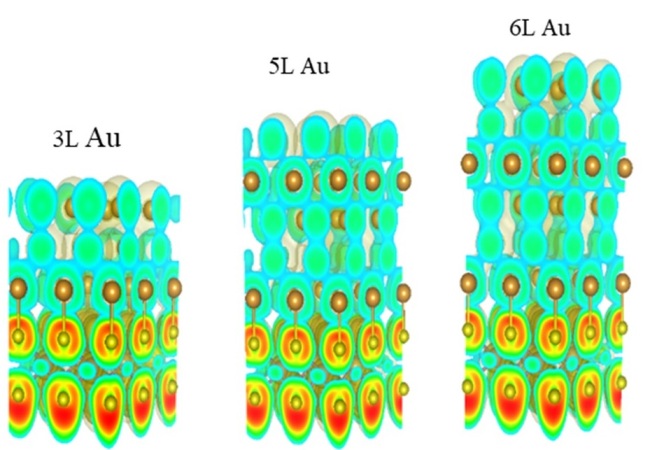
Abstract: A novel substrate consisting of a 2D MoS2/graphene van der Waals (vdW) heterostructure decorated with Au nanoparticles (AuNPs) was developed for surface enhanced Raman spectroscopy (SERS). A transfer-free chemical vapor deposition process was employed for layer-by-layer fabrication of graphene, followed with MoS2 directly on wafers of SiO2/Si without any metal catalyst. AuNPs were deposited on the MoS2/graphene via in situ electron-beam evaporation of Au at an elevated temperature in the range of 300-350 °C in high vacuum. Rhodamine 6G (R6G) was used as an SERS probe molecule and SERS sensitivity of 5x10-8 M using a non-resonance 633 nm laser, which is an order of magnitude higher than that reported on the AuNPs/graphene substrate using the same excitation. Higher SERS sensitivity of 5x10-10 M was obtained using a resonance 532 nm laser excitation. The observed SERS sensitivity enhancement can be attributed to the combination of the electromagnetic mechanism of the plasmonic AuNPs and the chemical mechanism of the AuNPs/MoS2/graphene vdW heterostructure via enhanced interface dipole-dipole interaction as compared to graphene or MoS2 only as suggested by a Density Function Theory calculation. Therefore, this AuNPs/MoS2/graphene vdW heterostructure is advantageous to practical applications in optoelectronics and biosensing.
_____________________________________________________
2019-3: Maogang Gong, Ridwan Sakidja, Ryan Goul, Dan Ewing, Matthew Casper,Alex Stramel, Alan Elliot, and Judy Z. Wu, "High-Performance All-Inorganic CsPbCl3 Perovskite Nanocrystal Photodetectors with Superior Stability", ACS Nano, 13, 2, 1772-1783, (2019).
Abstract: All-inorganic perovskites nanostructures, such as CsPbCl3 nanocrystals (NCs), are promising in many applications including photovoltaics, light-emitting diodes and photodetectors. Despite impressive performance demonstrated, a critical issue remains due to instability of the perovskites in ambient. Herein, we report a method of passivating crystalline CsPbCl3 NCs surface with 3-mercaptopropionic acid (MPA) and superior ambient stability is achieved. Printing of these colloidal NCs on the channel of graphene field effect transistors (GFETs) on solid SiO2/Si and flexible polyethylene terephthalate substrates was carried out to obtain CsPbCl3 NCs/GFET van der Waals heterojunction photodetectors for flexible and visible-blind ultraviolet detection at wavelength below 400 nm. Besides ambient stability, additional benefits of passivating surface charge trapping by the defects on CsPbCl3 NCs and facilitating high-efficient charge transfer between the CsPbCl3 NCs and graphene were provided by MPA. Extraordinary optoelectronic performance was obtained on the CsPbCl3 NCs/graphene devices including high ultraviolet responsivity exceeding 106 A/W, high detectivity of 2×1013 Jones, fast photoresponse time of 0.3 s, and ambient stability with less than 10% degradation of photoresponse after 2400 h. This result illustrates the critical importance of the perovskite NCs surface passivation not only to the performance but also the stability of the perovskite optoelectronic devices.
_____________________________________________________
2019-2: Samar Ghopry, Mohammed Alamri, Ryan Goul, Ridwan Sakidja and Judy Wu, Extraordinary Sensitivity of Surface-Enhanced Raman Spectroscopy of Molecules on MoS2 (WS2) Nanodomes/Graphene van der Waals Heterostructure Substrates ", Advanced Optical Materials, 1801249, (2019).
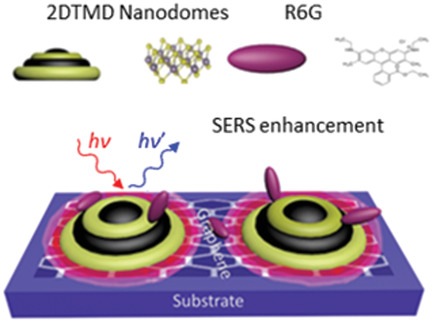
Abstract: Two-dimensional transition metal dichalcogenides (TMDs)/graphene van der Waals (vdW) heterostructures integrate the superior light-solid interaction in TMDs and charge mobility in graphene and therefore are promising for surface-enhanced Raman spectroscopy (SERS). This work reports a novel TMD (MoS2and WS2) nanodome/graphene vdW heterostructure SERS substrate on which an extraordinary SERS sensitivity has been achieved. Using fluorescent Rhodamine 6G (R6G) as probe molecules, the SERS sensitivity is in the range of 10-11-10-12 M on the TMD nanodomes/graphene vdW heterostructure substrates using 532 nm Raman excitation, which is comparable to the best sensitivity reported so far using plasmonic metal nanostructures/graphene SERS substrates, and is more than three orders of magnitude higher than that on the single-layer TMD and graphene substrates. Density functional theory simulation has revealed enhanced electric dipole moments and dipole-dipole interaction at the TMD/graphene vdW interface, yielding an effective means to facilitate an external electrostatic perturbation on the graphene surface and charge transfer. This not only promotes chemical enhancement on SERS, but also enables electromagnetic enhancement of SERS through excitation of localized surface plasmonic resonance on the TMD nanodomes. This TMD nanodome/graphene vdW heterostructure is therefore promising for commercial applications in high-performance optoelectronics and sensing.
_____________________________________________________
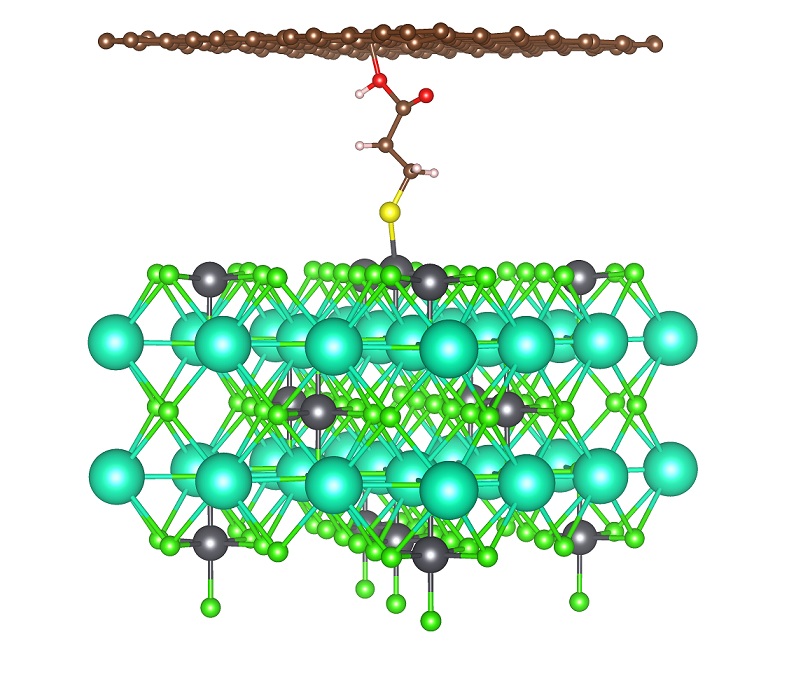
2019-1: Zhuolei Zhang, Ridwan Sakidja, Feng Hu, Beibei Xu, and Shenqiang Ren, "Self-Assembled Metal Molecular Networks by Nanoconfinement", The Journal of Physical Chemistry Letters, 10, 2, 206-213, (2019).
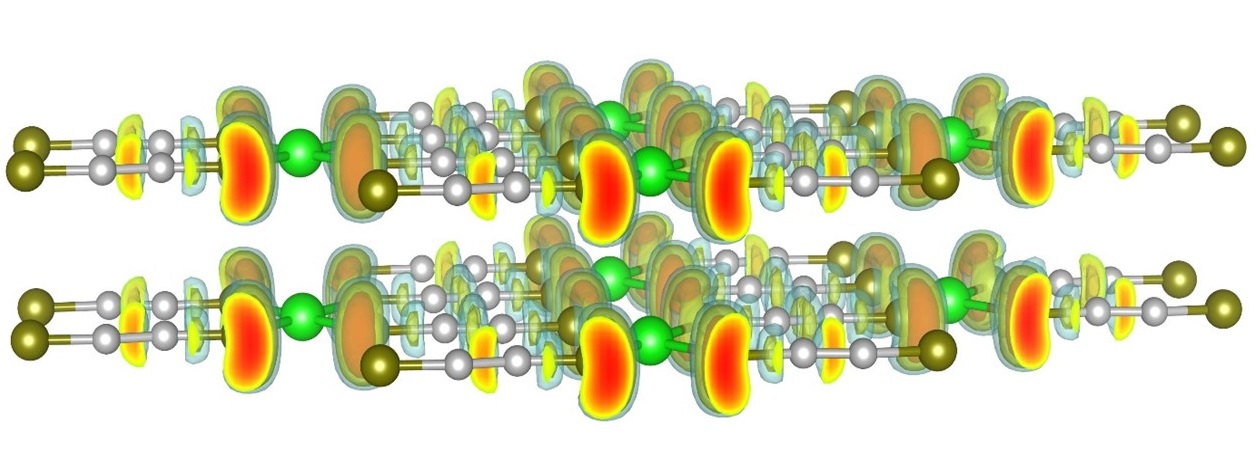
Abstract: Quasi-two-dimensional (2D) metal molecular networks (MMNs) often exhibit nanoconfinement effect and high degree of anisotropy, which are highly diverse in their mechanical, electronic, and magnetic functionalities. Here we report an interfacial self-assembly of mechanically robust 2D MMNs, in which 3d transition metals are interconnected via molecular thiol bridges. The Langmuir-Schäfer assembled freestanding 2D nanosheets exhibit highly desired anisotropic charge transport and spin susceptibility, in which light and magnetic field induced charge-transfer regulates its electronic interactions. Meanwhile, the mechanistic studies involving electronic structure reveals the molecular metal packing structure controlled nanoconfinement and charge transfer. This study opens the door to 2D ultrathin metal coordination nanostructures for emerging functional materials and devices.
2018
2018-5: Jamie Wilt, Ryan Goul, Jagaran Acharya, Ridwan Sakidja, and Judy Zhihong Wu, "In Situ Atomic Layer Deposition and Electron Tunneling Characterization of Monolayer Al2O3 on Fe for Magnetic Tunnel Junctions", AIP Advances, Vol. 8 [12], 125218, (2018).
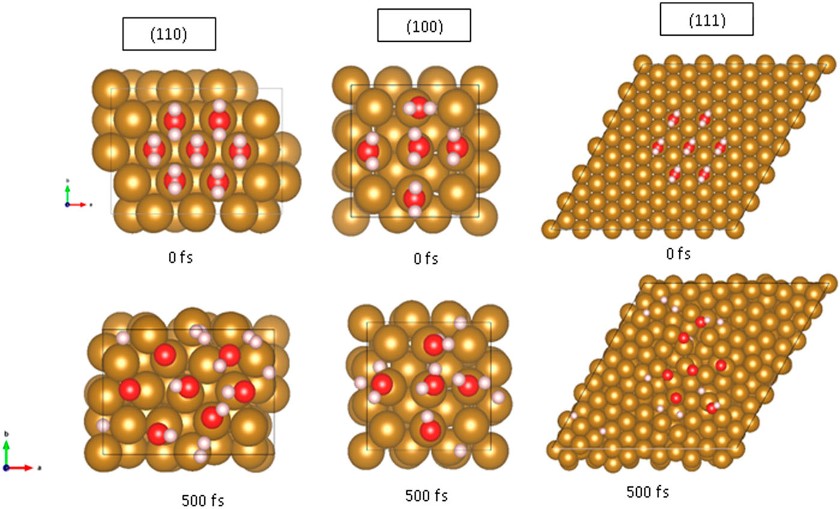
Abstract: Magnetic tunnel junctions (MTJs), formed through sandwiching an ultrathin insulating film (so-called tunnel barrier or TB), with ferromagnetic metal electrodes, are fundamental building blocks in magnetoresistive random access memory (MRAM), spintronics, etc. The current MTJ technology employs physical vapor deposition (PVD) to fabricate either amorphous AlOx or epitaxial MgO TBs of thickness around 1 nm or larger to avoid leakage caused by defects in TBs. Motivated by the fundamental limitation in PVD in, and the need for atomically thin and defect-free TBs in MTJs, this work explores atomic layer deposition (ALD) of 1-6 Å thick Al2O3 TBs both directly on Fe films and with an ultrathin Al wetting layer. In situ characterization of the ALD Al2O3 TB was carried out using scanning tunneling spectroscopy (STS). Despite a moderate decrease in TB height Eb with reducing Al wetting layer thicknesses, a remarkable Eb of ~1.25 eV was obtained on 1 Å thick ALD Al2O3 TB grown directly on an Fe electrode, which is more than twice of that of thermal AlOx TB (~0.6 eV). Achieving such an atomically thin low-defect TB represents a major step towards improving spin current tunneling in MTJs.
_____________________________________________________
2018-4: Md Delower Hossain, Robert Mayanovic, Sonal Dey, Ridwan Sakidja, Mourad Benamara, "Room-Temperature Ferromagnetism in Ni(II)-Chromia based Core-Shell Nanoparticles: Experiment and First Principles Calculations", Physical Chemistry Chemical Physics, Vol. 20[15], pp. 10396-10406, (2018).
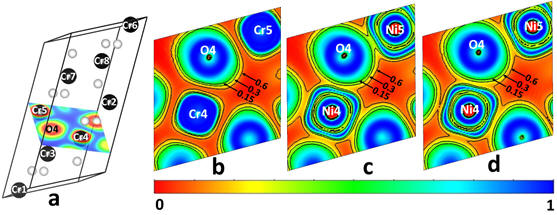
Abstract: We have synthesized bimagnetic core-shell nanoparticles containing a first-of-its-kind Ni(II)-chromia nanophase shell and a well-defined, epitaxial core-shell interface. Magnetic measurements reveal a substantial coercivity of the nanoparticles and a significant exchange bias effect between the antiferromagnetic chromia core and the ferromagnetic Ni(II)-chromia shell at low temperatures. The ferromagnetism and a weak exchange bias effect are found to persist to room temperature in the core-shell nanoparticles of ~ 57 nm average size. Our first principles Density Functional Theory (DFT) calculations confirm that the novel corundum-structued Ni(II)-chromia phase has an equilibrium cluster-localized ferromagnetic spin configuration. In addition, the DFT-based calculations show that the Ni(II)-chromia phase is a Mott-Hubbard insulator, with a narrowed energy band gap and increased covalent bonding due to strong hybridization between Ni 3d and O 2p levels in the upper portion of the valence band and within the band gap region. The antiferromagnetic, ferromagnetic and magnetoelectric properties of our core-shell nanoparticles make these well suited for patterned recording media and biomedical applications.
_____________________________________________________
2018-3: Maogang Gong, Ridwan Sakidja, Qingfeng Liu, Ryan Goul, Dan Ewing, Matthew Casper, Alex Stramel, Alan Elliot and Judy Z. Wu,"Broadband Photodetectors Enabled by Localized Surface Plasmonic Resonance in Doped Iron Pyrite Nanocrystals", Advanced Optical Materials, Vol. 6[8], Pages 1701241, (2018).
 |
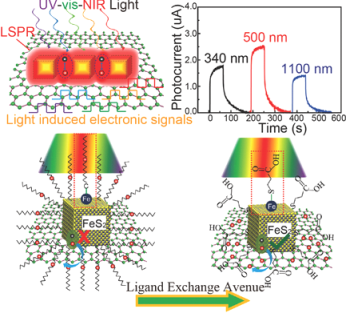 |
Abstract: The emerging capability to detect light over a broad spectral range is a key to technological applications in sensing, spectroscopy, imaging and communications. Colloidal semiconductor nanocrystal/graphene van der Waals heterojunctions provide a unique scheme that combines the spectral tunability and strong quantum confinement of the semiconductor nanocrystals sensitizers with superior charge mobility of graphene for extraordinary photoconductive gains. While high responsivity has been demonstrated, the spectral range is typically narrow limited by the cutoff of the semiconductor band gap of the nanocrystals. Here, a broadband photosensitizer is reported, based on doped Iron Pyrite nanocubes (FeS2 NCs) that exhibit strong localized surface plasmonic resonance (LSPR) spanning across ultraviolet through visible to near-infrared (UV–Vis–NIR). Using the printed LSPR FeS2 NCs/graphene van der Waals heterostructure, a broadband UV–Vis–NIR photoresponsivity in exceeding 1.08 × 106 A/W has been demonstrated through development of a ligand-exchange process to facilitate efficient charge transfer at the LSPR FeS2 NCs/graphene interface. This result demonstrates the viability of the LSPR semiconductor nanocrystal/graphene van der Waals heterostructure for high-performance broadband optoelectronics with scalability through direct printing.
_____________________________________________________
2018-2: Wesley Ng, Edwin Gnanakumar, Gadi Rothenberg, Erdni Batyrev, Sandeep Sharma, Pradeep Pujari, Heather Greer, Wuzong Zhou, Ridwan Sakidja, Michel Barsoum and N. R. Shiju,"Ti3AlC2 MAX-phase as an efficient catalyst for oxidative dehydrogenation of n-butane", Angewandte Chemie International Edition, 57 [6], 1485-1490 (2018).
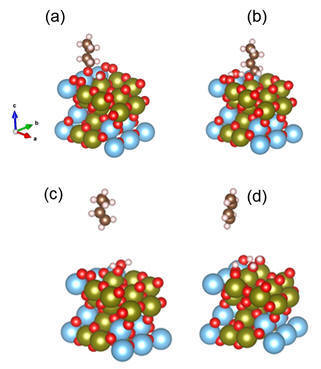
Abstract: Light alkenes are important raw materials for the synthesis of polymers and other products. Traditionally they are obtained from steam cracking and catalytic cracking. However, (oxidative) dehydrogenation of alkanes is gaining more importance to produce alkenes directly from natural gas. Here we report that Ti3AlC2, a MAX phase, which hitherto had not used in catalysis, efficiently catalyses the ODH of n-butane to butenes and butadiene, important intermediates for the synthesis of polymers and other compounds. The catalyst, which combines both metallic and ceramic properties, is stable for at least 30 h on stream, even at low O2:butane ratios without suffering from coking. This material has neither lattice oxygens nor noble metals, yet a unique combination of numerous defects and a thin surface Ti1-yAlyO2-y/2 layer that is rich in oxygen vacancies makes it an active catalyst. Given the large number of compositions available, MAX phases may find applications in several reactions.
_____________________________________________________
2018-1: Md. Delower Hossain, Robert Mayanovic, Ridwan Sakidja, Mourad Benamara and Richard Wirth, Magnetic properties of core-shell nanoparticles possessing a novel Fe(II)-chromia phase: an experimental and theoretical approach", Nanoscale, vol. 10, pp. 2138-2147 (2018).
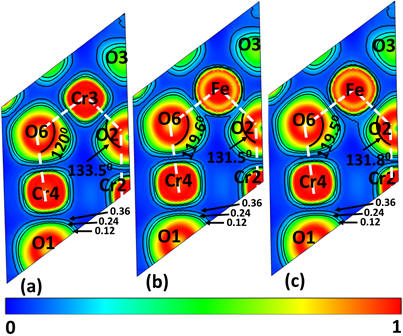
Abstract: Room-temperature ferrimagnetic and superparamagnetic properties, and the magnetic interactions between the core and shell, of our iron-incorporated chromia-based core shell nanoparticles (CSNs) have been investigated using a combination of experimental measurement and density functional theory (DFT) based calculations. We have synthesized CSNs having an epitaxial shell and well-ordered interface properties by utilizing our hydrothermal nanophase epitaxy (HNE) technique. The ferrimagnetic and superparamagnetic properties of the CSNs are manifested beyond room temperature and magnetic measurements reveal that the exchange bias interaction between the antiferromagnetic (AFM) core and ferrimagnetic (FiM) shell persists close to ambient temperature. The DFT calculations confirm the FiM ordering of the Fe-chromia shell. Our calculations show that the FiM ordering is associated with a band gap reduction, Fe-O d-p orbital hybridization, and AFM type Fe-Cr σ type superexchange interaction in the α-Fe0.40Cr1.60O2.92 shshell of the CSNs. The novel magnetic core-shell nanoparticles possess a shell comprised of a metastable Fe(II)-chromia phase, resulting in unique magnetic properties that make them ideal for magnetic device and medicinal applications.
2017
2017-7: Jamie Samantha Wilt, Ridwan Sakidja, Ryan Goul and Judy Z. Wu, "The effect of an interfacial layer on electron tunneling through atomically-thin Al2O3 tunnel barriers", ACS Applied Materials and Interfaces, 9 (42), pp 37468–37475 (2017).
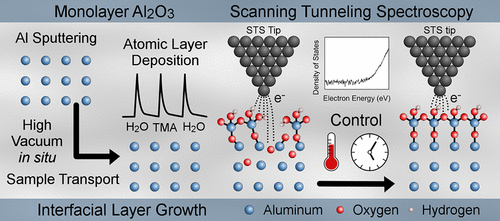
Abstract: Electron tunneling through high-quality, atomically thin dielectric films can provide a critical enabling technology for future microelectronics, bringing enhanced quantum coherent transport, fast speed, small size, and high energy-efficiency. A fundamental challenge is in controlling the interface between the dielectric and device electrodes. An interfacial layer (IL) will contain defects and introduce defects in the dielectric film grown atop, preventing electron tunneling through the formation of shorts. In this work, we present the first systematic investigation of the IL in Al2O3 dielectric films of 1-6 Å’s in thickness on an Al electrode. We integrated several advanced approaches: molecular dynamics to simulate IL formation, in situ high vacuum sputtering-atomic layer deposition (ALD) to synthesize Al2O3 on Al films, and in situ ultrahigh vacuum scanning tunneling spectroscopy to probe the electron tunneling through the Al2O3. The IL had a profound effect on electron tunneling. We observed a reduced tunnel barrier height and soft-type dielectric breakdown which indicate that defects are present in both the IL and in the Al2O3. The IL forms primarily due to exposure of the Al to trace O2 and/or H2O during the pre-ALD heating step of fabrication. As the IL was systematically reduced, by controlling the pre-ALD sample heating, we observed an increase of the ALD Al2O3 barrier height from 0.9 eV to 1.5 eV along with a transition from soft to hard dielectric breakdown. This work represents a key step towards the realization of high-quality, atomically thin dielectrics with electron tunneling for the next generation of microelectronics.
_____________________________________________________
2017-6: Soohyun Im, Michelle M. Paquette, Mohammed Belhadj-Larbi, Paul Rulis, Ridwan Sakidja and Jinwoo Hwang, Microscopy and Microanalysis, 23(S1), 1486-1487 (2017).
_____________________________________________________
2017-5:
Dayton G. Kizzire, Sonal Dey, Robert A. Mayanovic, Ridwan Sakidja, Kai Landskron, Manik Mandal, Zhongwu Wang, Mourad Benamara, "Studies of the Mechanical and Extreme Hydrothermal Properties of Periodic Mesoporous Silica and Aluminosilica Materials", Microporous and Mesoporous Materials, Vol. 252, pp. 69–78 (2017).
Abstract: In order to assess the suitability of mesoporous materials for applications in energy harvesting/storage processes occurring under extreme conditions, their mechanical, thermal and hydrothermal properties need to be fully investigated. In this study, the bulk mechanical and extreme hydrothermal properties of periodic mesoporous SBA-15 type silica and SBA-15 type aluminosilica (Al-SBA-15) were investigated using in situ small angle x-ray scattering (SAXS). In situ SAXS measurements were made on dry mesoporous SBA-15 silica and Al-SBA-15 aluminosilica samples as a function of pressure (at room temperature) to ~ 12 GPa and on the same mesoporous materials under extreme hydrothermal conditions (to 255 °C and ~ 114 MPa) using the diamond anvil cell (DAC). The analyses of the high-pressure SAXS data indicate that the mesoporous Al-SBA-15 aluminosilica has substantially greater bulk mechanical stability (isothermal bulk modulus k = 34.7(4.5) GPa) than the mesoporous SBA-15 silica (k = 12.0(3.0) GPa). Our molecular dynamics (MD) simulations are able to accurately model the bulk mechanical stability properties of mesoporous SBA-15 silica but underestimate the same properties of Al-SBA-15 aluminosilica. Analysis of the in situ SAXS data measured under extreme hydrothermal conditions indicates swelling of the pore walls due to water incorporation that is more significant in mesoporous Al-SBA-15 aluminosilica (~ 2x) than in SBA-15 silica. In addition, the Al-SBA-15 aluminosilica clearly exhibits superior hydrothermal stability compared to SBA-15 silica under the extreme experimental temperature and pressure conditions.
_____________________________________________________
2017-4: Md Shafiqul Islam, Paul Simanjuntak, Saibal Mitra and Ridwan Sakidja, "DFT Study on the Li Mobility in Li-Ion-Based Solid-State Electrolytes", MRS Advances, Vol. 2[54] (Energy Storage and Conversion), pp. 3277-3282 (2017).
Abstract: We have investigated the diffusion mechanisms of Li-ion in amorphous lithium phosphite (LiPO3) with an addition of sulphur. By applying the nudge elastic band (NEB)1 method in crystal LiPO3 and Li3PO4, we have confirmed the ease in diffusion pathways for Li ion in LiPO3 which is quite consistent with the previous theoretical finding. From our diffusion study in 0.5 Li2O- 0.5 P2O5 and 0.4 Li2SO4 – 0.6 (Li2O-P2O5) melts above 3000K investigated with ab-initio molecular dynamics (AIMD), as similarly has been confirmed by experiments at lower temperatures, we have demonstrated the benefit of S addition in increasing the Li+ mobility. We also found that the Li2SO4 additions into the glass result in a characteristic shift in Li-ion vibration to a lower vibrational frequency. In addition, the presence of oxygen surrounding the diffusion pathways appears to be essential in assisting the Li+ mobility in both crystalline and amorphous structures.
_____________________________________________________
2017-3: Jamie Wilt, Youpin Gong, Ming Gong, Feifan Su, Huikai Xu, Ridwan Sakidja, Alan Elliot, Rongtau Lu, Shiping Zhao, Siyuan Han, and Judy Z. Wu, "Atomically thin Al2O3 films for tunnel junctions ", Physical Review Applied, 7, 064022 (2017).
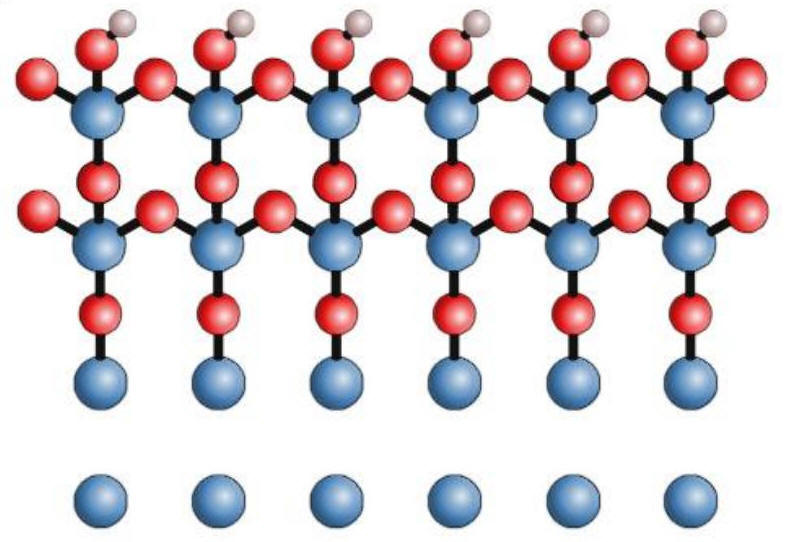
Abstract: Metal-insulator-metal tunnel junctions are common throughout the microelectronics industry. The industry standard AlOx tunnel barrier, formed through oxygen diffusion into an Al wetting layer, is plagued by internal defects and pinholes which prevent the realization of atomically thin barriers demanded for enhanced quantum coherence. In this work, we employ in situ scanning tunneling spectroscopy along with molecular-dynamics simulations to understand and control the growth of atomically thin Al2O3 tunnel barriers using atomic-layer deposition. We find that a carefully tuned initial H2O pulse hydroxylated the Al surface and enabled the creation of an atomically thin Al2O3 tunnel barrier with a high-quality M−I interface and a significantly enhanced barrier height compared to thermal AlOx. These properties, corroborated by fabricated Josephson junctions, show that atomic-layer deposition Al2O3 is a dense, leak-free tunnel barrier with a low defect density which can be a key component for the next generation of metal-insulator-metal tunnel junctions.
_____________________________________________________
2017-2: Anthony Ascone and Ridwan Sakidja, "MDM2 case study: Computational protocol utilizing protein flexibility improves ligand binding mode predictions ", International Journal of Computational Biology and Drug Design, Vol. 10 No. 3 pp. 217 - 224 (2017).
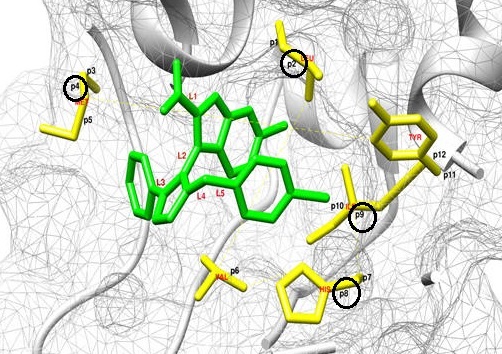
Abstract: Recovery of the P53 tumor suppressor pathway via small molecule inhibitors of onco-protein MDM2 highlights the critical role of computational methodologies in targeted cancer therapies. Molecular docking programs in particular, have become essential during computer-aided drug design by providing a quantitative ranking of predicted binding geometries of small ligands to proteins based on binding free energy. In this study, we found improved ligand binding mode predictions of small medicinal compounds to MDM2 based on RMSD values using AutoDock and AutoDock Vina employing protein binding site flexibility. Additional analysis suggests a data mining protocol using linear regression can isolate the particular flexible bonds necessary for future optimum docking results. The implementation of a flexible receptor protocol based on a priori knowledge obtained from data mining will improve accuracy and reduce costs of high throughput virtual screenings of potential cancer drugs targeting MDM2.
_____________________________________________________
2017-1: Mohammad Delower Hossain, Robert A. Mayanovic, Ridwan Sakidja, Mourad Benamara,"An experimental and theoretical study of the optical, electronic and magnetic properties of novel inverted a-Cr2O3@a-Mn0.35Cr1.65O2.94 core shell nanoparticles", Journal of Materials Research, Volume 32, Issue 2, pp. 269-278,(2017).
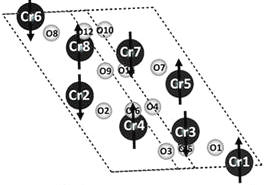
Abstract: Magnetic core–shell nanoparticles (CSNs) have potential applications in spintronic devices, drug delivery systems, and magnetic random access memory. By use of our hydrothermal nano-phase epitaxy method, we have accomplished synthesis of novel, well-ordered α-Cr2O3@α-Mn0.35Cr1.65O2.94 inverted CSNs. XRD and TEM analyses show a core–shell structure with corundum phase throughout the core and shell with a minimal amount of interface defects. TEM-EDX and XPS data show Mn having the +2 oxidation state in the shell of the CSNs. Magnetization measurements at 5 K show a weak coercivity (HC) value of 8 Oe and an exchange bias field (HE) of 293 Oe. Ab-initio calculations show that Mn incorporation in α-Cr2O3 results in narrowing of the energy band gap, substantiated by UV–Vis measurements, and half metallic behavior in case of Mn(III) substitution. Our calculations substantiate that Mn substitution in α-Cr2O3 results in a combination of antiferromagnetic and weak ferrimagnetic character of our CSNs.
2016
2016-4:
Batu Hunca, Chamila Dharmawardhana, Ridwan Sakidja, and Wai-Yim Ching, "Ab initio calculations of thermomechanical properties and electronic structure of vitreloy Zr41.2Ti13.8Cu12.5Ni10Be22.5", Physical Review B, 94, 144207 (2016).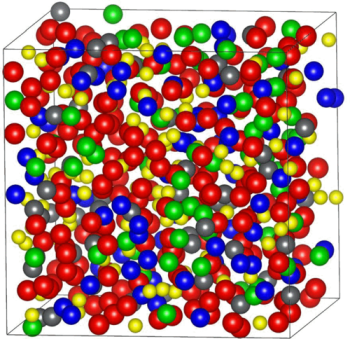
Abstract: The thermomechanical properties and electronic structure of vitreloy (Zr41.2Ti13.8Cu12.5Ni10Be22.5) are investigated using accurate ab initio molecular dynamic (AIMD) simulations and ab initio calculations. The structure of the model with 512 atoms is validated by comparison to the experimental data with calculated thermomechanical properties in good agreement with the existing measurements. Detailed calculation of the electronic structure and bonding at the density functional level is obtained. It is revealed that the traditional definition of bond length in metallic glasses has a limited interpretation, and any theory based on geometrical consideration of their values for discussion on the structural units in metallic glasses has similarly limited applications. On the other hand, we advocate the use of a quantum mechanical based metric, the total bond order density (TBOD), and their partial components or PBOD as valuable parameters to characterize the interatomic bonding in multicomponent glasses such as vitreloy.
_____________________________________________________
2016-3: Dayton G. Kizzire, James Thomas, Sonal Dey, Hayley Osman, Robert A. Mayanovic, Ridwan Sakidja, Zhongwu Wang, Manik Mandal and Kai Landskron, "Investigations of the mechanical and hydrothermal stabilities of SBA-15 and Al-SBA-15 mesoporous materials", MRS Advances, Volume 1, Issue 35 (Materials Design), pp. 2453-2458, (2016).
Abstract: Quasi-two-dimensional (2D) metal molecular networks (MMNs) often exhibit nanoconfinement effect and high degree of anisotropy, which are highly diverse in their mechanical, electronic, and magnetic functionalities. Here we report an interfacial self-assembly of mechanically robust 2D MMNs, in which 3d transition metals are interconnected via molecular thiol bridges. The Langmuir-Schäfer assembled freestanding 2D nanosheets exhibit highly desired anisotropic charge transport and spin susceptibility, in which light and magnetic field induced charge-transfer regulates its electronic interactions. Meanwhile, the mechanistic studies involving electronic structure reveals the molecular metal packing structure controlled nanoconfinement and charge transfer. This study opens the door to 2D ultrathin metal coordination nanostructures for emerging functional materials and devices.
_____________________________________________________
2016-2: Maogang Gong, Ridwan Sakidja, and Shenqiang Ren, "Composition and oxidation controlled magnetism in ternary FeCoNi nanocrystals", Nano Research, Vol. 9 [3], Pages 831-836, (2016)
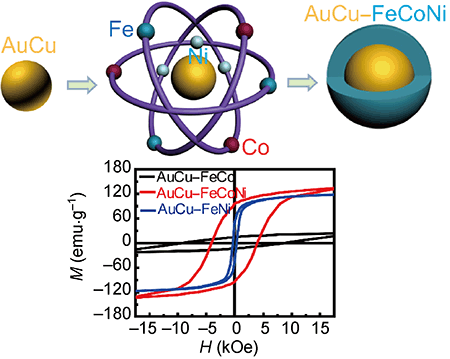
Abstract: Ternary FeCoNi metallic nanostructures have attracted significant attention due to their high saturation magnetization, unique mechanical properties, and large corrosion resistance. In this study, we report a controlled synthesis of ternary FeCoNi nanocrystals using solution-based epitaxial core–shell nanotechnology. The thickness and stoichiometry of the FeCoNi nanocrystals affect their magnetic characteristics, which can be controlled by a phase transformation-induced tetragonal distortion. Furthermore, surface oxidation of the stoichiometry-controlled FeCoNi nanostructures can drastically enhance their magnetic coercivity (up to 8,881.6 Oe for AuCu–FeCo), and optimize the AuCu–FeCo0.8Ni0.2 performance corresponding to the saturated magnetization of 134.4 emu·g−1 and coercivity of 4,036.7 Oe, which opens the possibility of developing rare-earth free high energy nanomagnets.
_____________________________________________________
2016-1: Sonal Dey, Sean Anderson, Robert A. Mayanovic, Ridwan Sakidja, Kai Landskron, Berenika Kokoszka, Manik Mandal, Zhongwu Wang, "Experimental and theoretical investigation of a mesoporous KxWO3 material having superior mechanical strength", Nanoscale, Vol. 8, Pages 2937-2943, (2016).
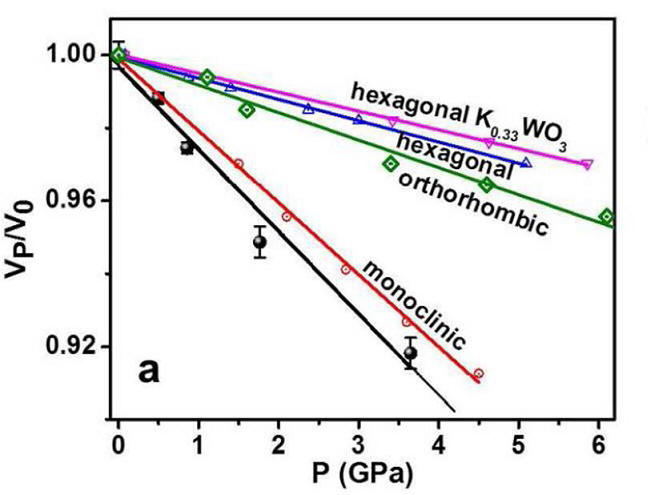
Abstract: Mesoporous materials with tailored properties hold great promise for energy harvesting and industrial applications. We have synthesized a novel tungsten bronze mesoporous material (KxWO3; x ~ 0.07) having inverse FDU-12 type pore symmetry and a crystalline framework. In situ small angle X-ray scattering (SAXS) measurements of the mesoporous K0.07WO3 show persistence of a highly ordered meso-scale pore structure to high pressure conditions (~18.5 GPa) and a material with remarkable mechanical strength despite having ~35% porosity. Pressure dependent in situ SAXS measurements reveal a bulk modulus κ = 44 ± 4 GPa for the mesoporous KxWO3 which is comparable to the corresponding value for the bulk monoclinic WO3 (γ-WO3). Evidence from middle angle (MAXS) and wide angle X-ray scattering (WAXS), high-resolution transmission electron microscopy (HR-TEM) and Raman spectroscopy shows that the presence of potassium leads to the formation of a K-bearing orthorhombic tungsten bronze (OTB) phase within a monoclinic WO3 host structure. Our ab-initio molecular dynamics calculations show that the formation of the OTB phase provides superior strength to the mesoporous K0.07WO3.
2015
2015-6: Maogang Gong, Xin Jin, Ridwan Sakidja, and Shenqiang Ren,
"Synergistic strain engineering effect of hybrid plasmonic, catalytic and magnetic core-shell nanocrystals," Nano Letters, Vol. 12[15], Pages 8347-8353, (2015).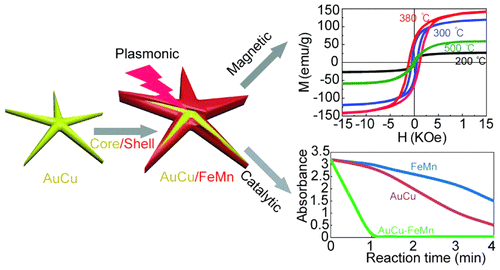
Abstract: Hybrid core–shell nanocrystals, consisting of distinct components, represent an emerging functional material system, which could facilitate synergistic coupling effects via integrating drastically different functionalities. Here we report a unique strain engineering effect induced by phase transformation between plasmonic core and magnetic shell materials, which leads to a facile surface reconstruction of bimetallic core–shell nanocrystals to enhance their synergistic magnetic and catalytic properties. This advancement dramatically results in two orders of magnitude enhancement in magnetic coercivity and significant improvement in catalytic activity. Mechanistic studies involving the kinetic measurement and theoretical modeling uncover a structural distortion and surface rearrangement mechanism during the core–shell phase transformation pathway. This facile methodology could potentially open up the new design of multifunctional artificial hybrid nanostructures by the combination of phase transformation and surface engineering for emerging technological applications.
_____________________________________________________
2015-5: W. Qin, M. Gong, X. Chen, T. A. Shastry, R. Sakidja, G. Yuan, M. C. Hersam, M. Wuttig and S. Ren, Advanced Materials, Vol. 27, Pages 734-739 (2015).
Abstract: A new type of carbon charge-transfer magnet, consisting of a fullerene acceptor and single-walled carbon nanotube donor, is demonstrated, which exhibits room temperature ferromagnetism and magnetoelectric (ME) coupling. In addition, external stimuli (electric/magnetic/elastic field) and the concentration of a nanocarbon complex enable the tunabilities of the magnetization and ME coupling due to the control of the charge transfer.
_____________________________________________________
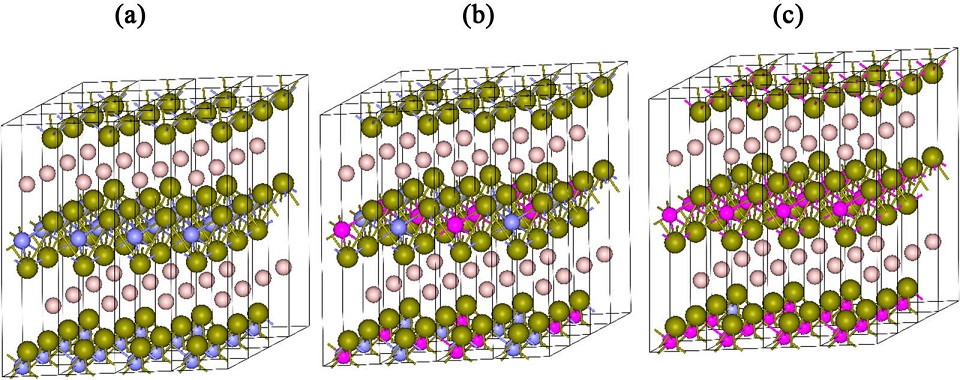
2015-4: Sitaram Aryal, Ridwan Sakidja, Lizhi Ouyang and Wai-Yim Ching, "Elastic and electronic properties of Ti2Al(CxN1-x) ) solid solutions", Journal of the European Ceramic Society, Vol. 35[12], Pages 3219-3227 (2015).
Abstract: The elastic coefficients and mechanical properties (bulk modulus, shear modulus, Young's modulus and Poisson's ratio) of Ti2Al(CxN1−x) ) continuous solid solutions for x from 0 to 1 are calculated using ab initio DFT methods on 4 × 4 × 1 supercell models. It is shown that the properties of these solid solutions do not vary linearly with x. Although the lattice constant c is almost constant for x ≤ 0.5, a increases linearly. For x > 0.5, c starts to increase with x while the rate of increase in a slows down. For x between 0.5 and 0.85, the elastic coefficients and the mechanical parameters show interesting dependence on x and crossovers, signifying the complex interplay in the structure and properties in Ti2Al(CxN1−x)solid solutions. The nonlinear variations in mechanical properties are explained in terms of subtle difference in the electronic structure and bonding between nitrides and carbides in complex MAX phase compounds.
_____________________________________________________
2015-3: Chandra Dhakal, Sitaram Aryal, Ridwan Sakidja, Wai-Yim Ching, "Approximate lattice thermal conductivity of MAX phases at high temperature", Journal of the European Ceramic Society, Vol. 35[12], Pages 3203-3212 (2015).
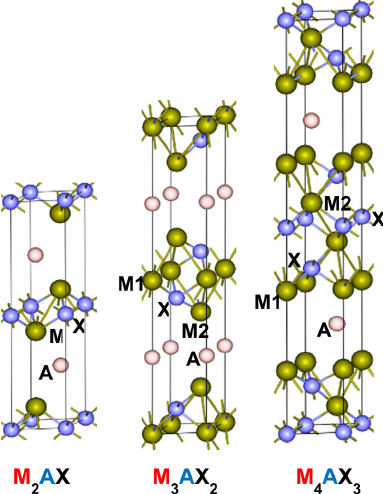
Abstract: The temperature dependent lattice thermal conductivity (κph) of MAX phases, Mn+1AXn are calculated using the Debye theory as outlined by Slack. At high temperature the formula derived by Slack is a reasonable approximation to estimate the lattice thermal conductivity. The calculation used the large data base of elastic coefficients of stable MAX phases established recently. It is found that MAX phases with “A” = Al have higher κph at 1300 K, and the majority of MAX carbides have higher κph than MAX nitrides. We have also calculated the minimum thermal conductivities of these MAX phases using the empirical formula suggested by Clarke. It is shown that the minimal lattice thermal conductivities of MAX carbides and nitrides are closer to each other in the 211 phases than in higher n phases. The calculated κph for 8 MAX phases at 1300 K are in reasonable agreement with experimental data, especially in Ti2AlC, Nb4AlC3, Ta4AlC3, Nb2AlC and Nb2SnC phases.
_____________________________________________________
2015-2: Qingfeng Liu, Youpin Gong, Jamie Samantha Wilt, Ridwan Sakidja, Judy Wu, "Synchronous growth of AB-stacked bilayer graphene on Cu by simply controlling hydrogen pressure in CVD process", Carbon, Vol. 93, Pages 199–206 (2015).
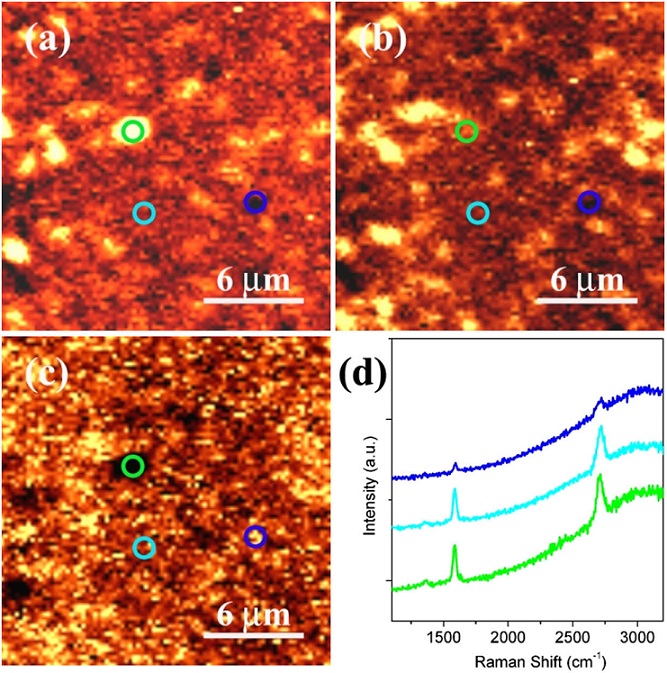
Abstract: AB-stacked bilayer graphene has attracted considerable attention due to its feasibility of band gap tuning. Although synthesis of bilayer graphene on Cu has been reported using chemical vapor deposition (CVD) through a layer-by-layer growth mechanism, the process is long and complicated due to lack of catalytic assistance of Cu to the second graphene layer growth. Here we show that theoretical modeling demonstrates an alternative synchronous growth of bilayer graphene on Cu is possible by passivating the top graphene nuclei edges with hydrogen to allow carbon diffusion underneath the top graphene nuclei for bottom graphene layer formation. Moreover, such a growth mechanism has been achieved experimentally in a facile CVD method by simply controlling the H2 pressure. Bilayer graphene with high coverage of over ~ 95% and a high AB stacking ratio of up to ~ 90% has been obtained within a short growth time of 30 min. Also, graphene with single, double and multiple layers can be obtained by simply controlling the hydrogen pressure. This result represents the demonstration of the fast synchronous AB-stacked bilayer graphene growth, which is important to scalable manufacture of graphene with controllable layer number and stacking required for practical applications.
_____________________________________________________
2015-1: C. C. Dharmawardhana, Ridwan Sakidja, Sitaram Aryal, Wai-Yim Ching,"In search of zero thermal expansion anisotropy in Mo5Si3 by strategic alloying", J. Alloys and Compounds , Vol. 620, Pages 427-433 (2015).
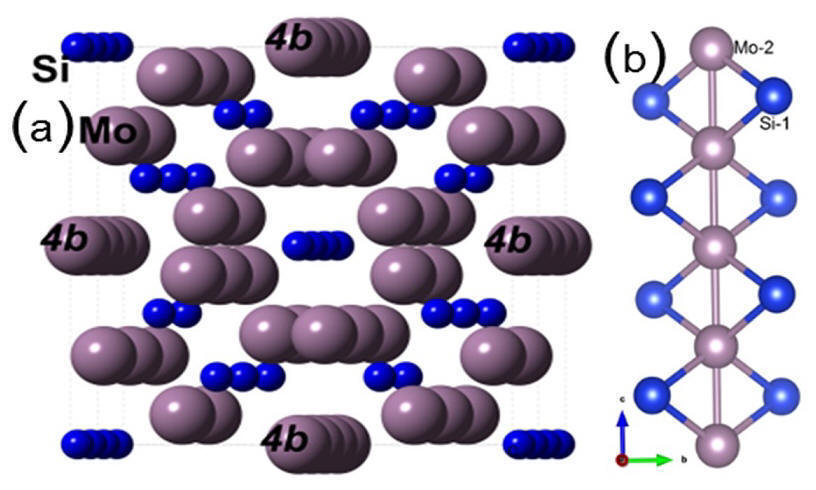
Abstract: Reducing the thermal expansion anisotropy (TEA) of alloy compounds is one of the most important issues for their potential applications in high temperature environment. The Mo5Si3 (T1 phase) is known to be an important intermetallic compound with high melting temperature. Unfortunately, its large TEA renders it unsuitable for high temperature structural/coating applications. Many attempts have been made in the past to reduce TEA by substituting Mo by other transition metal ions such as V with little success and some unexpected observations. Here we use accurate ab initio molecular dynamics (AIMD) simulations to obtain the TEA from thermal expansion coefficients for two T1 phase alloy systems (Mo,V)5Si3 and Mo5(Si,Al)3. We demonstrate that strategic alloying with Al substituting Si can achieve zero TEA for T1 phase. The microscopic origin of this outstanding thermomechanical properties in this alloy is explained by the calculation of higher order elastic constants in conjunction with atom and direction-resolved phonon density of states.
2014
2014-7: Sitaram Aryal, Ridwan Sakidja, M. W. Barsoum and Wai-Yim Ching, "A genomic approach to the stability, elastic, and electronic properties of the MAX phases", Physica status solidi (b),Vol. 251, Issue 8, Pages 1480–1497 (2014).
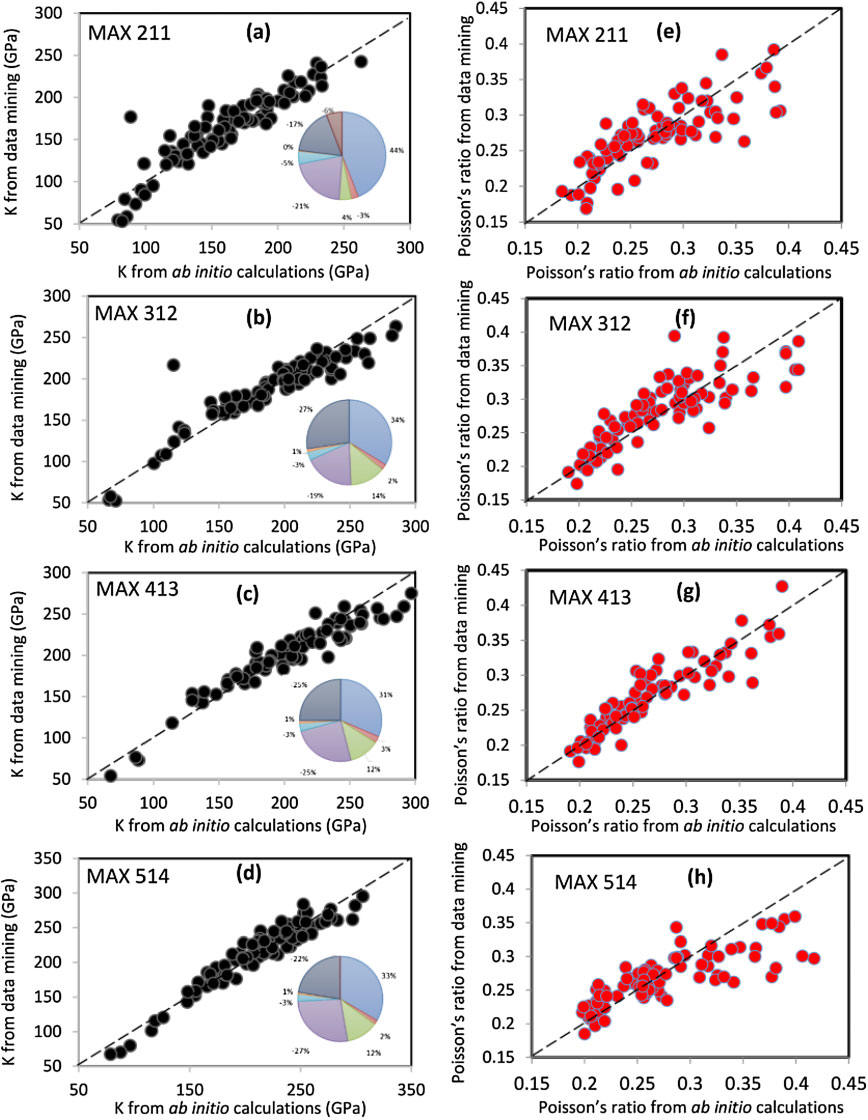
Abstract: In this study, we report a comprehensive assessment on the elastic and electronic properties of 792 possible MAX (Mn+1AXn) ) phases with n = 1–4 using ab initio methods. These crystals are then screened based on their elastic and thermodynamic stability resulting in a large database of 665 viable crystals. All the experimentally verified MAX phases passed the screening. Various correlations among and between them are fully explored. In particular, the key elements in the interdependence between the elastic properties together with mechanical parameter derived from them and the electronic structure are identified. Detailed analysis of various correlation plots shows that there is a clear correspondence between bulk modulus K and total bond order density (TBOD). Calculations show a marked difference between the carbides and nitrides. This database is also used to test the efficacy of data mining algorithms for materials genome. We further identified several thermodynamically stable new MAX phases with unusual mechanical parameters that have never been synthesized in the laboratory or theoretically investigated. The complete database on the elastic and electronic structure together with the mechanical parameters for these 665 MAX phases compounds are included in the Supplementary Materials and fully accessible.
_____________________________________________________
2014-6: Neng Li, Ridwan Sakidja, Sitaram Aryala, Wai-Yim Ching, "Densification of a continuous random network model of amorphous SiO2 glass", Phys. Chem. Chem. Phys., Vol. 16, Pages 1500-1514 (2014).
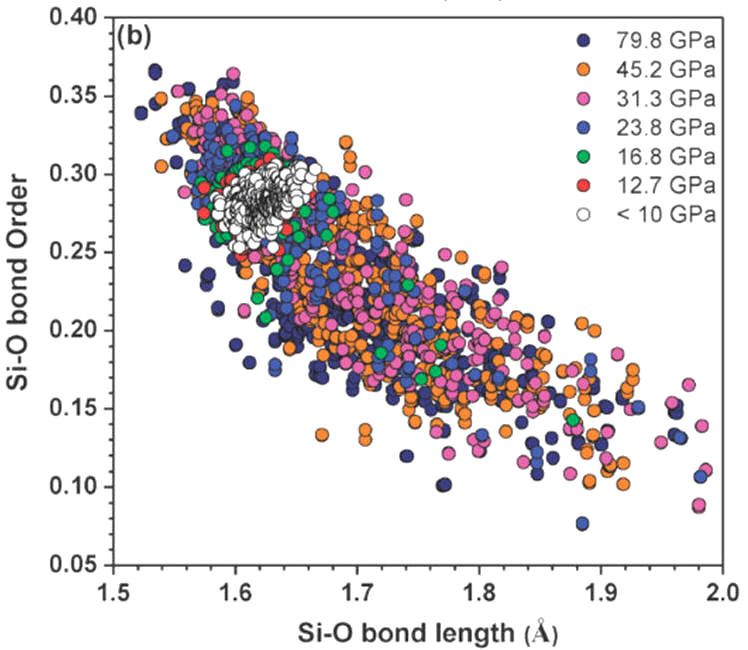
Abstract: We have investigated the mechanism of densification of a nearly perfect continuous random network (CRN) model of amorphous SiO2 (a-SiO2) ) glass with 1296 atoms and periodic boundary conditions. The model has no under- or over-coordinated atoms and small bond length and bond angle distributions. This near-perfect model is systematically densified up to a pressure of 80 GPa using ab initio constant-pressure technique. By assessing a full spectrum of properties including atomic structure, bonding characteristics, effective charges, bond order values, electron density of states, localization of wave functions, elastic and mechanical properties, and interband optical absorption at each pressure, we reveal the pertinent details on the structural, mechanical and optical characteristics of the glass model under pressure. They all confirm the central theme that amorphous to amorphous phase transformation (AAPT) from a low-density state to a high-density state is at a pressure between 20 and 35 GPa in this nearly ideal a-SiO2 network. This pressure range represents an upper limit for such a transition in vitreous silica. The phase transformation roots from the change of Si–O bonding from a mixture of ionic and covalent nature at low pressure to a highly covalent bonding under high pressure. In addition, the calculated theoretical refractive index of the glass model as a function of the pressure is reported for the first time and in good agreement with the available experimental data.
_____________________________________________________
2014-5: Amelia Bengtson, Hyo On Nam, Saumitra Saha, Ridwan Sakidja, Dane Morgan, "First-principles molecular dynamics modeling of the LiCl–KCl molten salt system", Computational Materials Science, Vol. 83, 15, Pages 362–370 (2014).
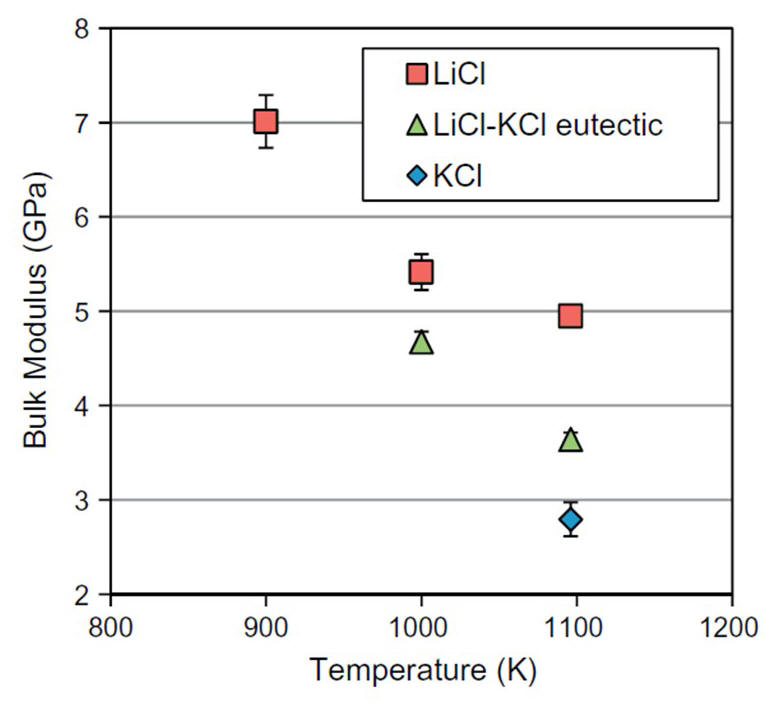
Abstract: Properties of molten salts are of interest for a wide-range of applications, including nuclear waste partitioning, heat transfer fluids, and synthesis methods. While there has been extensive work showing the value of molecular modeling with interatomic potentials to predict molten salt properties there have been very limited studies of molten salts from a fully first-principles approach. In order to establish optimal approaches and their strengths and limitations in first-principles molten salt modeling, this work provides extensive first-principles molecular dynamics simulations of the LiCl–KCl molten salt system that are validated against existing literature. The basic thermokinetic properties of volume, thermal expansion, bulk modulus, and diffusivity, are calculated for LiCl, KCl and the eutectic LiCl–KCl liquids at multiple temperatures. Convergence testing reveals 216-atom unit cells and simulation times of 6–12 ps are sufficient to provide results with acceptable uncertainties and agreement with experimental data. The results provide a framework of first principles molecular dynamics simulations in the LiCl–KCl molten salt system that can be extended in future research to predict less well-established properties, e.g., the behavior of solutes.
_____________________________________________________
2014-4: Hyo On Nam, Amelia Bengtson, K. Votler, Saumitra Saha, Ridwan Sakidja, Dane Morgan, "First-principles molecular dynamics modeling of the molten flouride salt with Cr solute", J. Nuclear Materials, Vol. 449 [1-3], 15, Pages 148–157 (2014).
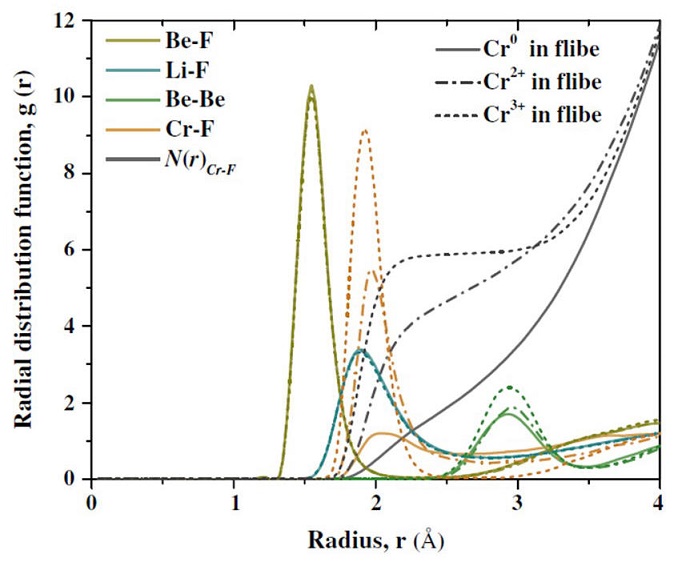
_____________________________________________________
2014-3: Neng Li, Ridwan Sakidja and Wai-Yim Ching,"Ab initio study on the adsorption mechanism of oxygen on Cr2AlC (0001) surface", Applied Surface Science, Vol. 315, Pages 45-54 (2014).
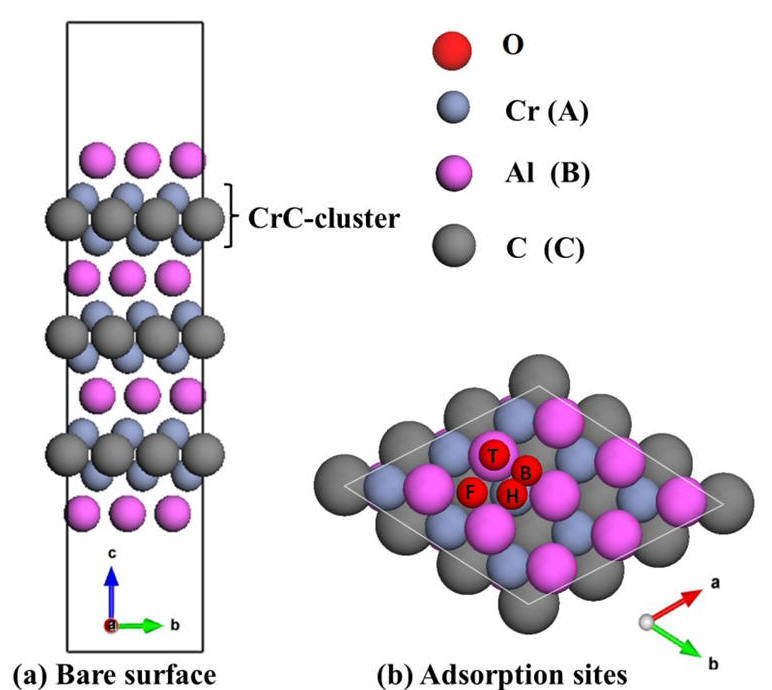
Abstract: We performed an investigation of the early stage oxidation of the technologically relevant Cr2AlC (0 0 0 1) surface by ab initio calculations. In this work, the geometrical structure, bonding character, and electronic structure of the Cr2AlC (0 0 0 1) bare surface with or without single oxygen atom and molecular adsorption had been systematically investigated by DFT calculation. Four possible terminations Al-, Cr(C)-, C- and Cr(Al)-terminated Cr2AlC (0 0 0 1) surface are considered. The corresponding surface energies of the four configurations are compared, indicating that the Al- and Cr(C)-terminated surfaces are more stable than the other two cases C- and Cr(Al)-terminated surfaces. For the most stable case, Al-terminated Cr2AlC (0 0 0 1) surface, a detailed model describing the oxygen–surface interactions is developed by assessing the adsorption energetics of various adsorption mechanisms. Based on the evaluation of the energetics and the structural properties of the atomistic models generated, the results point to a consistent picture of the initial stage of the Cr2AlC (0 0 0 1) surface oxidation resembling that of (1 1 1) layer of pure Al FCC phase.
_____________________________________________________
2014-2: I. P. Downs, J. H. Perepezko, R. Sakidja and S. R. Choi, "Suppressing CMAS attack with a MoSiB-based coating", Surface and Coatings Technology, Vol. 239, Pages 138 -146 (2014).
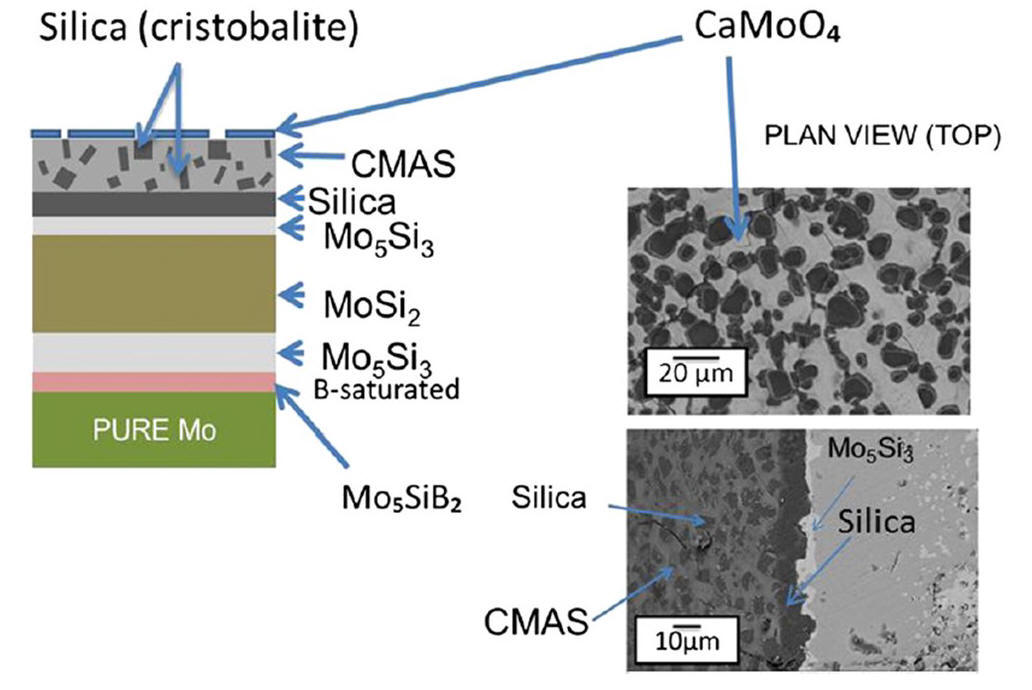
Abstract: Molten CMAS (calcia–magnesia–aluminosilica) deposits penetrate and interact with thermal barrier coatings (TBCs) in gas-turbine engines to degrade the zirconia-based TBCs and the overall TBC performance. To address the CMAS attack some approaches have been advanced based upon sealing the TBC or altering the TBC composition. A new strategy is presented based upon the application of a MoSiB based coating applied by pack cementation. The interaction between the MoSiB-based coating on a Mo substrate and CMAS is evaluated on two types of natural CMAS sands and on a commonly studied synthetic CMAS. The MoSiB-based coating arrests CMAS melt penetration by an in-situ reaction that yields crystallization products that act to immobilize the CMAS. The coating design is demonstrated to be effective in preventing CMAS attack during isothermal exposure at temperatures up to at least 1500 °C and during thermal cycling.
_____________________________________________________
2014-1: R. Sakidja, J. H. Perepezko and P. Calhoun, "Synthesis, Thermodynamic Stability and Diffusion Mechanism of Al5Fe2-based coatings", Oxidation of Metals, Vol. 81 [1], Pages 167-177 (2014).

Abstract: Aluminide coating of steels enables more efficient power generation through higher operating temperatures. Low-temperature (T < 660 °C) pack cementation aluminide coatings form an Al5Fe2 phase which allows for the development of a large Al flux, but the mechanism is not clear. The coating structures and resultant oxides were examined in both austenitic and ferritic steels at 1,000 and 800°C to evaluate the high temperature oxidation behavior in air. To understand the relatively fast Al diffusion, the stability of the Al5Fe2 phase and the defect structure have been examined by a cluster expansion method with density functional theory calculations. The Al5Fe2 phase has a low site occupancy and a high vacancy content that promotes rapid kinetics. The high vacancy concentration in the Al5Fe2 phase can be traced to the interaction between Al and vacancies along the [001] chains. The analysis offers useful guidance to enable an effective control of low temperature aluminizing.